the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 17 Nov 2025
| 17 Nov 2025
γ effects identify preferentially populated rotamers of CH2F groups: side-chain conformations of fluorinated valine analogues in a protein
Elwy H. Abdelkader
Nicholas F. Chilton
Ansis Maleckis
Gottfried Otting
Using cell-free protein synthesis, the protein G B1 domain (GB1) was prepared with uniform high-level substitution of valine by (2S,3S)-4-fluorovaline, (2S,3R)-4-fluorovaline or 4,4'-difluorovaline. The 19F nuclear magnetic resonance (NMR) signals are distributed over a wide spectral range. The fluorinated samples maintain the relative 1H chemical shifts of the wild-type protein, opening a convenient route to assign the 19F-NMR signals. For the singly fluorinated residues, the 13C chemical shifts of the remaining CH3 group are subject to a γ effect that depends on the population of different rotameric states of the CH2F group and correlates with 3JFC coupling constants. In addition, the preferentially populated rotamers are reflected by the γ-gauche effect on 19F chemical shifts, which correlates with 3JHF couplings. Some of the side-chain conformations determined by these restraints position the fluorine atom near a backbone carbonyl group, a non-intuitive finding that has previously been observed in the high-resolution crystal structure of a different protein. Through-space scalar 19F–19F couplings due to transient fluorine–fluorine contacts are observed between residues 39 and 54.
- Article
(4616 KB) - Full-text XML
-
Supplement
(2526 KB) - BibTeX
- EndNote




Fluorine atoms incorporated into proteins provide convenient probes for monitoring and analysis by 19F nuclear magnetic resonance (NMR) spectroscopy. As CF and CH groups in organic compounds feature notably similar spatial requirements and hydrophobicities, the global substitution of a single amino acid type in a protein by a selectively fluorinated analogue is usually possible with minimal structural perturbation and limited penalty with regard to fold stability. If the fluorine atom is installed in a methyl group, the resulting CH2F group has the freedom to respond to the increased spatial requirement of the fluorine atom by preferential population of those rotamers that are most readily accommodated by the chemical environment. Recently, we showed that the 19 kDa protein E. coli peptidyl–prolyl cis-trans isomerase B (PpiB), which contains five leucine residues, can be produced with high-level uniform substitution of leucine for (2S,4S)-5-fluoroleucine (FLeu1), (2S,4R)-5-fluoroleucine (FLeu2), or 5,5'-difluoroleucine (diFLeu) by using cell-free protein synthesis (Tan et al., 2024). As demonstrated by X-ray crystal structures, the structural perturbations caused by these amino acid substitutions were minimal (Frkic et al., 2024a). The 19F-NMR signals were dispersed over a large chemical shift range and acted as highly sensitive probes of ligand binding (Tan et al., 2024). Similarly, when PpiB was produced with (2S,3S)-4-fluorovaline (Fig. 1), the melting temperature of the protein decreased by no more than 15 °C despite the substitution of 16 valine residues, and the crystal structure revealed a fully conserved protein fold. The spectral range of the 19F-NMR spectrum exceeded 20 ppm (Frkic et al., 2024b).
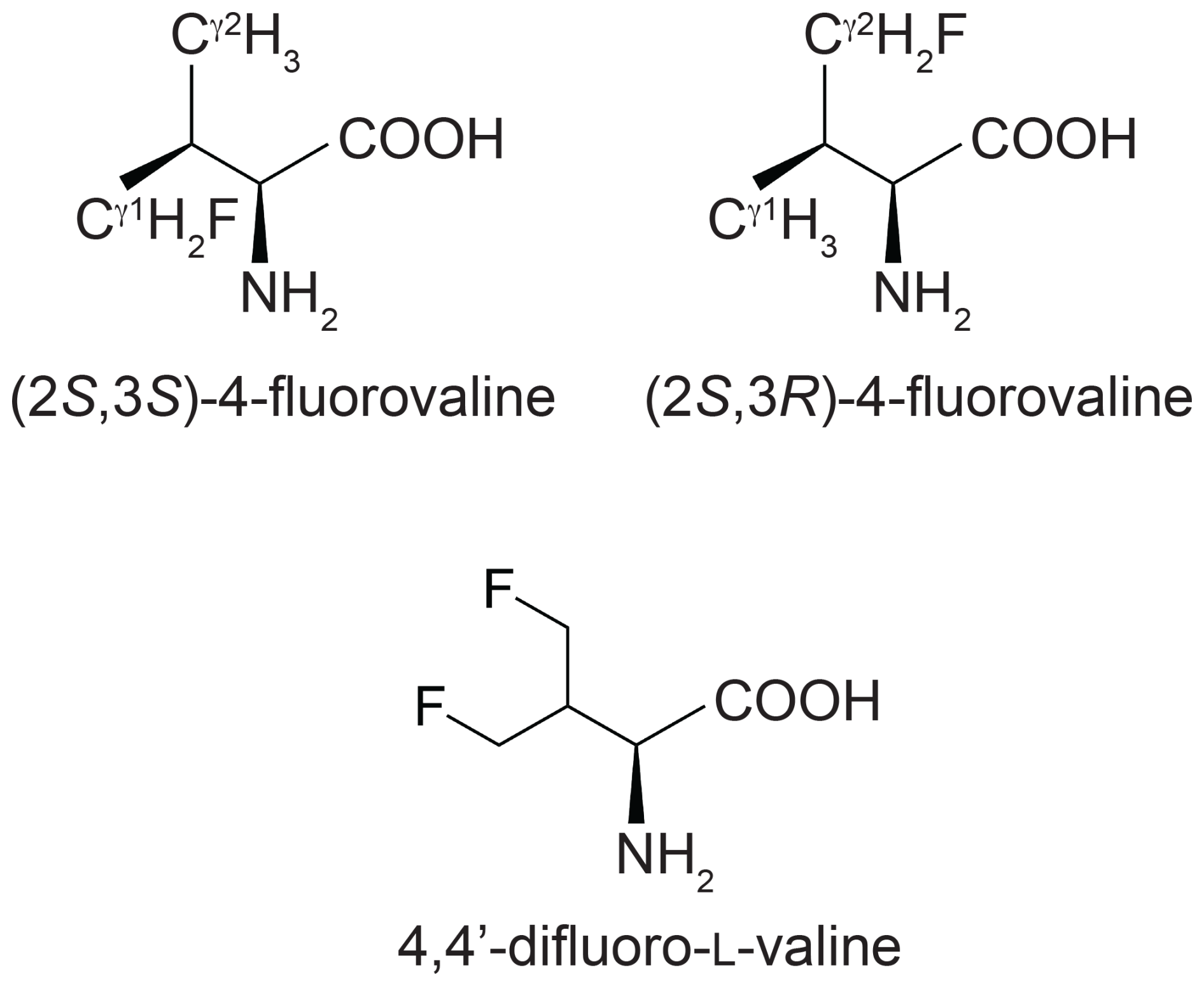
Figure 1Chemical structures of the fluorinated valine analogues used in the present work. (2S,3S)-4-fluorovaline, (2S,3R)-4-fluorovaline and 4,4'-difluoro-L-valine are referred to in the following as FVal1, FVal2 and diFVal, respectively. Samples of GB1 made with uniform substitution of valines for FVal1, FVal2 and diFVal are referred to as GB1-1, GB1-2 and GB1-d, respectively.
Among the light elements with spin , fluorine is unusual in that contacts between two 19F spins readily produce through-space scalar 19F–19F couplings, TSJFF (Hierso, 2014), which can also be observed in proteins (Kimber et al., 1978; Orton et al., 2021). In GB1, made with fluorinated leucine analogues instead of canonical leucine, TSJFF couplings between CδH2F groups are readily manifested in [19F,19F]-TOCSY spectra, although the couplings are small (up to about 3 Hz; Tan et al., 2025). In agreement with a significant energy barrier between staggered rotamers calculated for 1-fluoropropane (Feeney et al., 1996), the crystal structures of PpiB made with fluorinated leucine and valine residues show that the CH2F groups strongly prefer one of the three staggered rotamer conformations while frequently populating more than single rotamers. As TSJFF couplings depend on short-range orbital overlap (Hierso, 2014), the small size of TSJFF couplings may signal transient fluorine–fluorine contacts between non-uniformly rotating CH2F groups.
To gain a better understanding of the preferential populations of different rotamer conformations of CH2F groups under solution conditions at room temperature, we produced GB1 with fluorinated valine residues. Figure 1 shows the fluorinated valine analogues used, which were incorporated by producing GB1 by means of cell-free protein synthesis from a mixture of the 20 amino acids with canonical valine substituted by one of the fluorinated analogues. The results show that (i) the γ effect of 13C chemical shifts (Tonelli and Shilling, 1981; Tonelli et al., 1982; Fürst et al., 1990; Günther, 2013) provides a readily accessible restraint to define the preferential rotamers populated by a CH2F group, (ii) multiple rotamers are usually populated, (iii) there appears to be a bias towards rotamers positioning the fluorine atom near the positively polarised carbon of a carbonyl group, and (iv) TSJFF couplings can be observed between neighbouring fluorovaline residues. The results highlight the potential of fluorinated amino acids as NMR probes that perturb protein structure minimally.
2.1 Protein production, stability and NMR samples
The preparation of the proteins GB1-1, GB1-2 and GB1-d and their analysis by mass spectrometry have been described previously (Maleckis et al., 2022). Subsequent preparations used commercial FVal1 and FVal2 from Enamine (Ukraine). The respective proteins were made by means of cell-free protein synthesis replacing valine with either FVal1, FVal2 or diFVal at 2 mM concentration. Mass spectrometry analysis of the intact proteins showed that the most abundant species was the protein where every valine was substituted by the fluorinated analogue. A significant fraction featured one canonical valine residue among the four valine sites in the protein, amounting to about 30 % of the main species in GB1-1 and GB1-2 but 80 % in the case of GB1-d, indicating that increasing fluorination challenges the recognition of the amino acids by the E. coli valyl–tRNA synthetase. In the case of GB1-d, random incorporation of one canonical valine together with three diFVal residues would thus yield four different species, each about 5-fold less abundant than the main GB1-d product where all valine residues are replaced by diFVal.
Monitoring the heat denaturation of the proteins by circular dichroism revealed melting temperatures Tm between 67 and 70 °C, i.e. about 10 °C lower than for the wild-type protein under the same conditions (Fig. S1 in the Supplement; Tan et al., 2025).
The NMR samples were prepared in 90 % H2O/10 % D2O, 100 mM NaCl and 20 mM MES buffer at pH 6.5 and 0.1 mM trifluoroacetate as the calibration standard (−75.25 ppm). The protein concentration of the final NMR samples was about 0.7 mM (1.6 mM for HOESY spectra of GB1-1 and GB1-2).
2.2 1D 19F-NMR
The 1D 19F-NMR resonances of GB1-1 and GB1-2 are distributed over a large chemical shift range and are well resolved, except for a single instance of signal overlap in GB1-1, and all eight expected 19F-NMR signals are resolved in GB1-d (Fig. 2). The greater chemical heterogeneity of the GB1-d sample is evidenced by additional peaks of about 20 % intensity (Fig. 2c). In previous work on GB1 produced with diFLeu, the 19F chemical shifts were found to depend strongly on whether nearby leucine sites in the protein contain canonical leucine or the fluorinated analogue (Tan et al., 2025). Therefore, the low-intensity peaks in the 19F-NMR spectrum of GB1-d most likely originate from the species with one valine residue and three diFVal residues. As two of the valine sites are close to each other in GB1, a diFVal residue in one of the sites and a valine residue in the other explains why four of the weak 19F-NMR signals in GB1-d are well resolved.
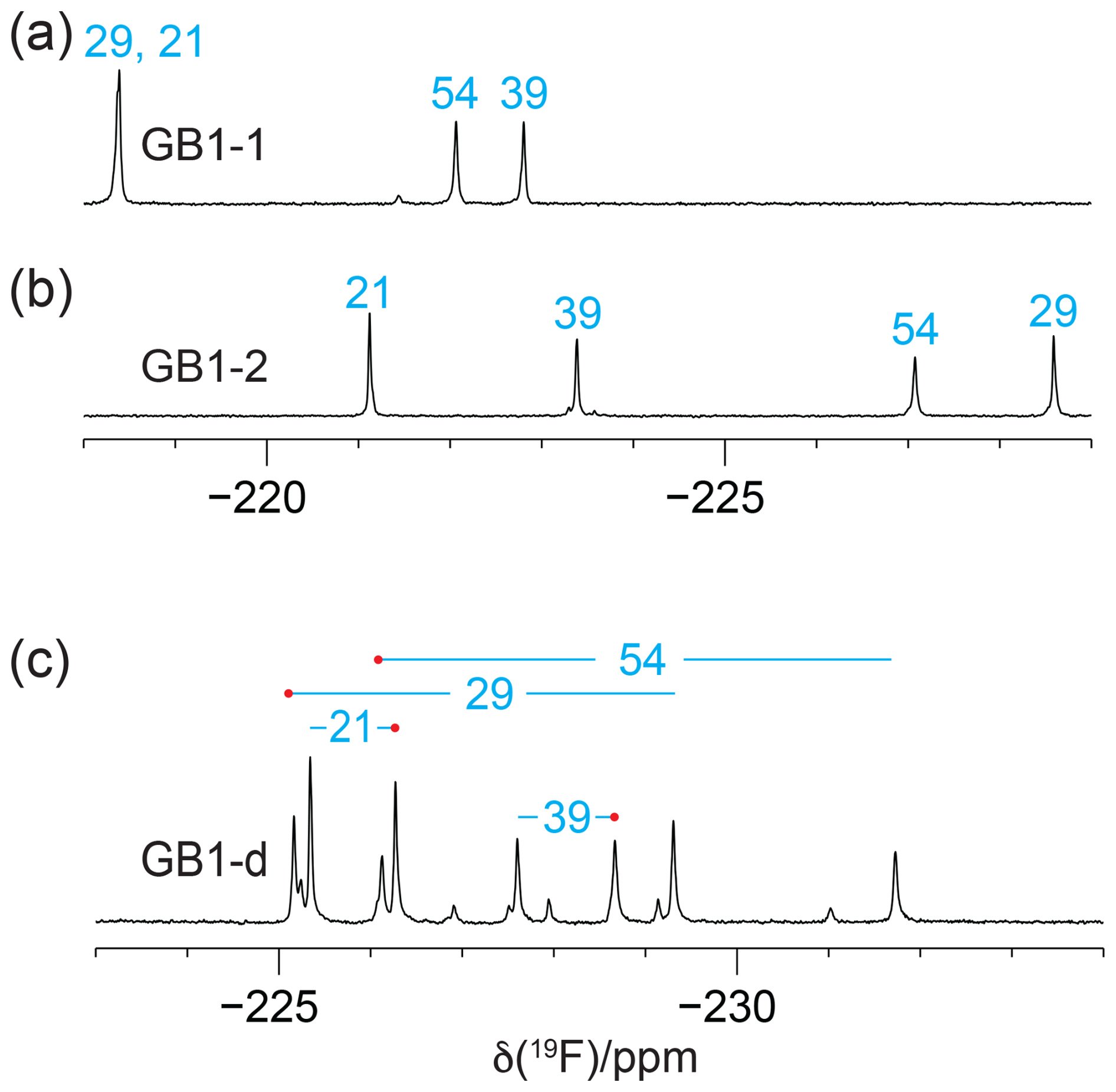
Figure 2The 1D 19F-NMR spectra of GB1 made with fluorinated valine analogues. Unless stated otherwise, all NMR spectra in this work were recorded at 25 °C on a 400 MHz NMR spectrometer with 1H decoupling. The peaks are labelled with the sequence-specific resonance assignments (blue). (a) GB1 made with FVal1. (b) GB1 made with FVal2. (c) GB1 made with diFVal. Red dots identify the resonances assigned to the Cγ1H2F groups.
19F-NMR spectra recorded without 1H decoupling display broad multiplets. Fundamentally, each CγH2F group produces a triplet due to splittings by the 2JHF coupling constant (about 47 Hz), which is split further by the 3JHF coupling with the Hβ atom. If the 3JHF coupling is of similar magnitude to the 2JHF coupling, the multiplet appears like a quartet. An example is the 19F-NMR signal of the γ2-fluorine of residue 54 (Fig. 3b and c). In contrast, the γ1-fluorine of this residue appears to be a triplet of doublets, indicating a much smaller 3JHF coupling constant (Fig. 3a). The inverse correlation of 3JHF couplings with 19F chemical shifts is a manifestation of the γ-gauche effect predicted by quantum calculations (Tonelli et al., 1982; Feeney et al., 1996) and is experimentally confirmed for fluorinated leucine residues in the proteins PpiB (Tan et al., 2024; Frkic et al., 2024a) and GB1 (Tan et al., 2025) and for FVal1 in PpiB (Frkic et al., 2024b). In the case of GB1 with fluorinated valine residues, the correlation between three-bond couplings and 19F chemical shifts is less striking. For example, residue 29 in GB1-2 produces the most high-field shifted resonance, but the multiplet indicates a smaller three-bond coupling than for residue 54 (Fig. 3b). As the 19F chemical shifts depend on the rotamers populated by the CH2F groups, as well as their chemical environment, the 19F chemical shifts observed in GB1-d do not simply recapitulate those of GB1-1 and GB1-2, necessitating independent resonance assignment strategies.
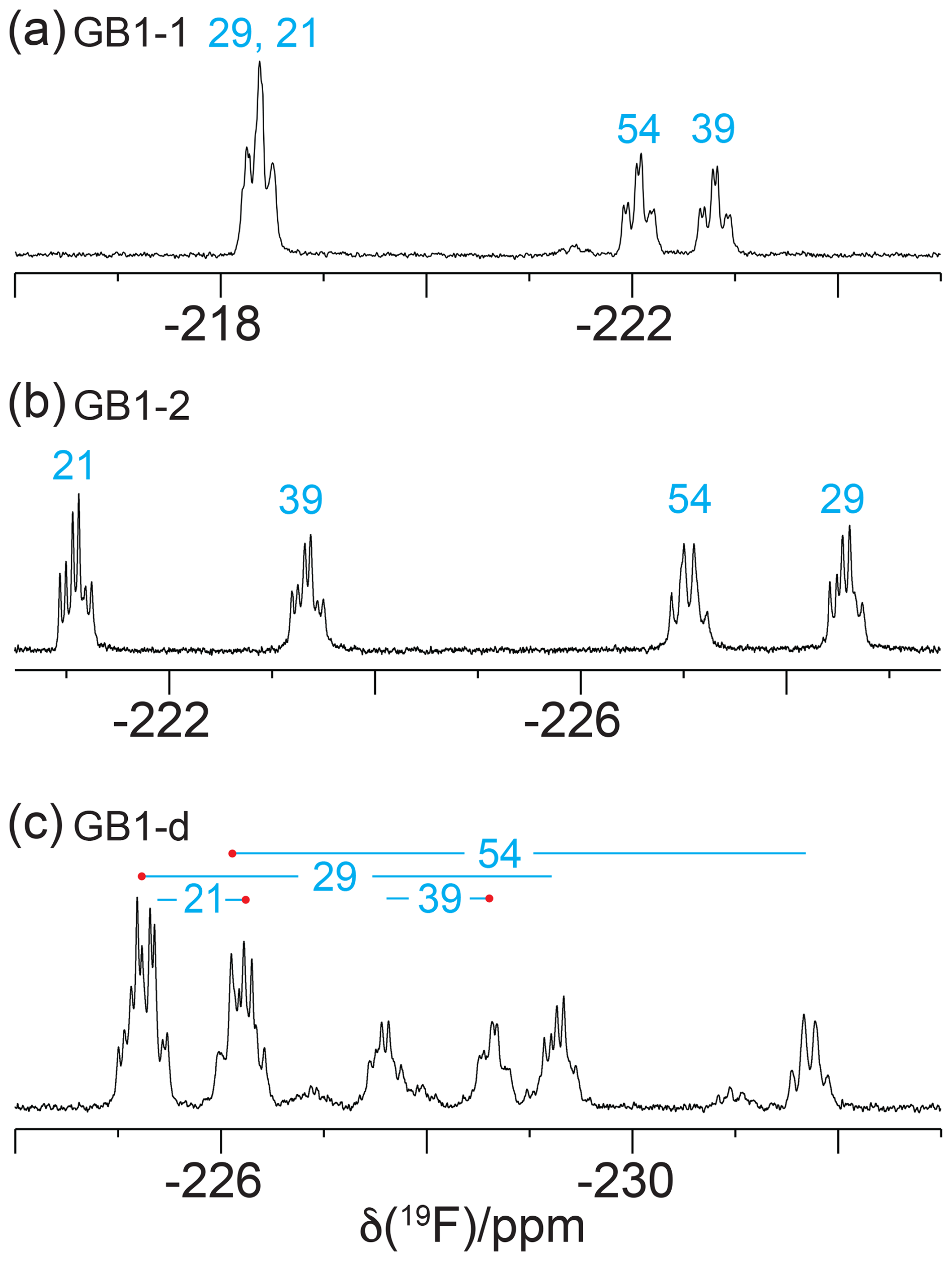
Figure 3The 1D 19F-NMR spectra recorded without 1H decoupling, using the same samples and conditions as in Fig. 2.
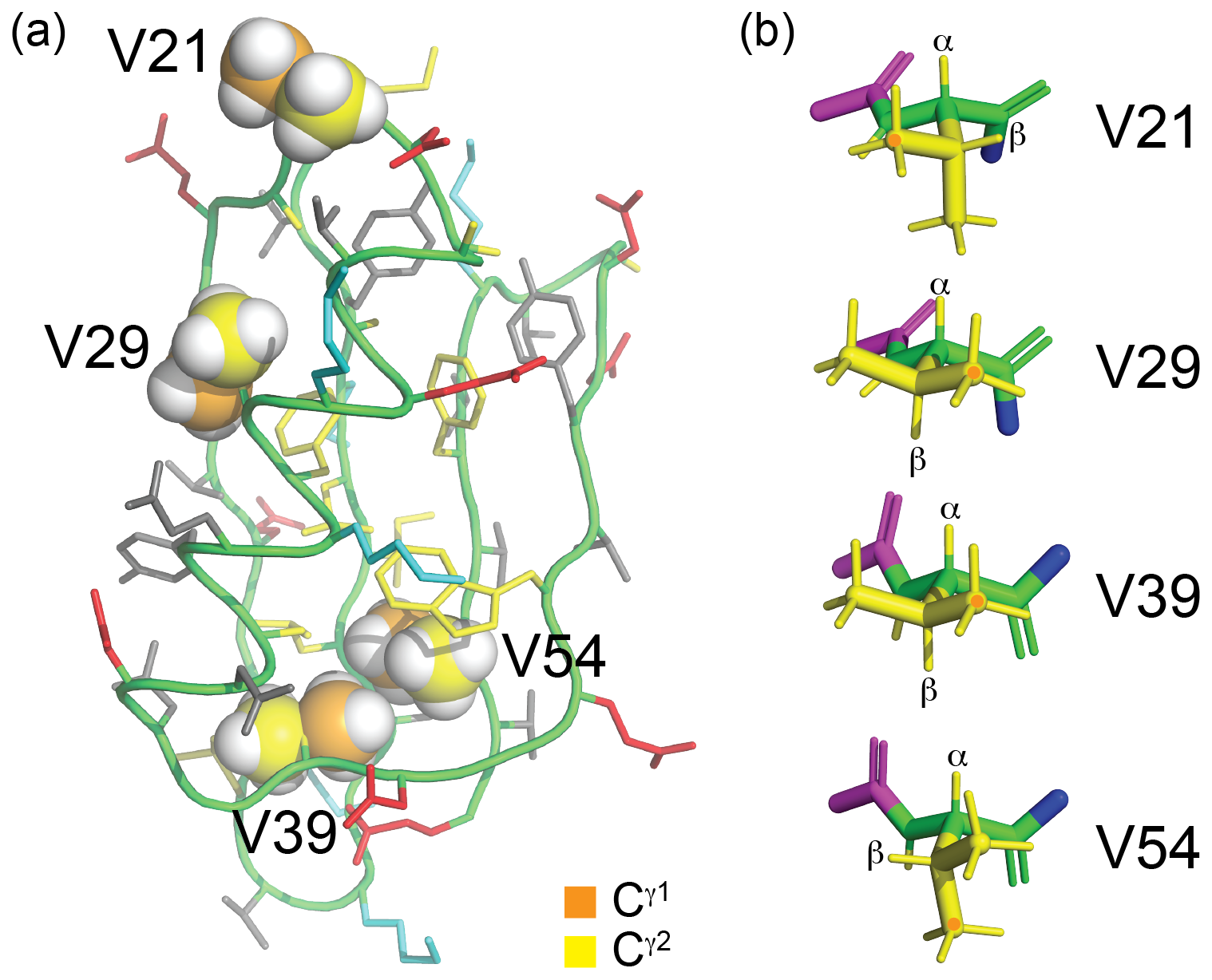
Figure 4Solution structure of GB1 (PDB ID 3GB1, Juszewski et al., 1999). (a) Overview showing the methyl groups of the four valine residues in a space-filling representation. The γ1 and γ2-carbon atoms are coloured orange and yellow, respectively. V39 and V54 are buried, whereas V29 is partially solvent-exposed, and V21 is highly solvent-exposed. The side chains of the other amino acids are coloured blue (positively charged), red (negatively charged), grey (hydrophilic) and yellow (hydrophobic). (b) Stick representation of the four valine residues, showing the backbone N, Cα, C′ and O atoms in green and the side chain in yellow. The nitrogen of the previous residue is shown in blue, and the Cα, C′ and O atoms of the following residue are shown in magenta. The rotamer states of the methyl groups, which vary between different conformers in the solution structure, were rotated to depict the ideal staggered conformations. The Hα and Hβ atoms are labelled, and the Cγ1H3 groups are marked with an orange dot. In three of the valine residues, the valine carbonyl carbon is close to a methyl hydrogen, involving the Cγ1H3 group of residues 29 and 39 and the Cγ2H3 group of residue 54.
Inversion–recovery experiments of GB1-d indicated T1(19F) relaxation times ranging between 0.35 s (residue 39) and 0.6 s (residue 54), i.e. slightly longer than for GB1 with difluoroleucine, where T1(19F) was about 0.3 s (Tan et al., 2025). R1ρ(19F) measurements of GB1-d ranged between 9 s−1 (γ2-fluorine of residue 21) and 26 s−1 (γ2-fluorine of residue 54; Table S2 in the Supplement). On average, these rates are faster than those of diFLeu residues previously studied in the same protein (Tan et al., 2025), which may be attributed to the greater proximity of the CH2F groups of diFVal to the protein backbone, thus limiting side-chain mobility. The broadest signals were observed for the most deeply buried residues, indicating that the peak heights are sensitive indicators of the side-chain mobilities (Fig. 2).
2.3 Resonance assignments
The sequence-specific resonance assignments of the 1H-NMR spectra of GB1-1, GB1-2 and GB1-d were readily obtained by conventional 2D NMR spectroscopy, using NOESY (200 ms mixing time), TOCSY (80 ms mixing time) and DQF-COSY experiments recorded on an 800 MHz NMR spectrometer. Structural conservation was indicated by conserved chemical shifts. For example, ring currents induced by W43 cause characteristic high-field shifts of the Hβ and Cγ2H3 resonances of V54 (to about −0.3 and 0.4 ppm, respectively), and these high-field shifts persist in the samples made with fluorinated valine analogues (Fig. S2 in the Supplement). Specifically, all fluorinated valine analogues appear to maintain the side-chain conformations of the wild-type protein as indicated by the intensities of their Hα–Hβ COSY cross-peaks, which were intense for residues 29 and 39 (indicative of a dihedral angle χ1 of about 180°) but weak or below the level of detection for residues 21 and 54 (as expected for a small 3J(Hα,Hβ) coupling), in accordance with the conformations shown in Fig. 4b.
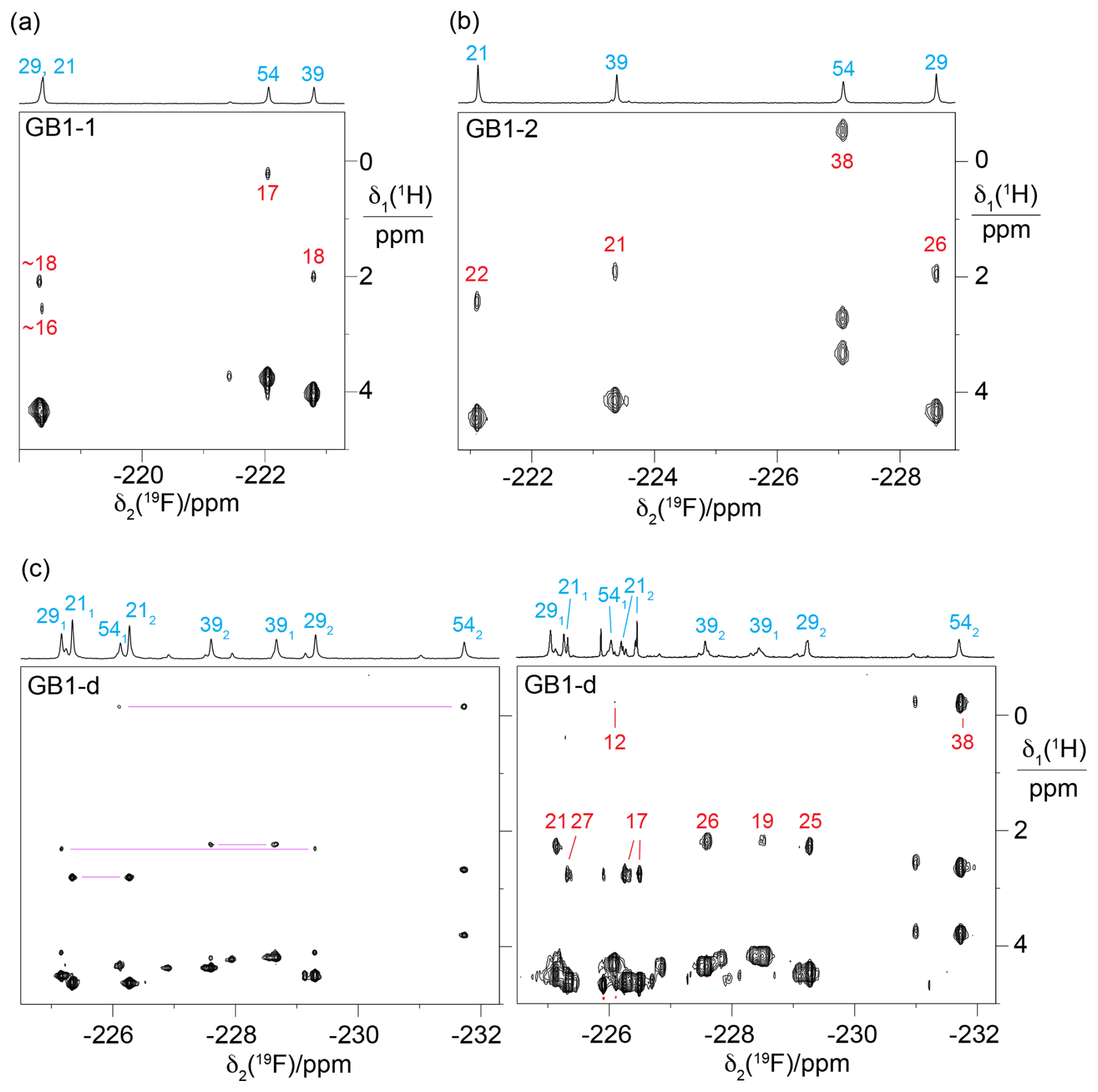
Figure 5Short-delay 1H,19F correlation experiments (Tan et al., 2024) for measuring 3JHF couplings and the 19F-detected [1H,1H]-TOCSY spectrum for assigning the 19F-NMR signals belonging to the same diFVal residue. All spectra were recorded on a 400 MHz NMR spectrometer with a room-temperature probe, except for the short-delay 1H,19F correlation experiment of GB1-d, which was recorded on a 500 MHz NMR spectrometer equipped with a cryoprobe. The short-delay 1H,19F correlation experiments were conducted with the delays Δ and δ set to 7 and 2.5 ms, respectively. The 1D 19F-NMR spectra are shown at the top together with the resonance assignments. The 1H chemical shifts of the CH2F groups are between 2.5 and 5 ppm. The cross-peaks with the 1Hβ resonances are labelled with the 3JHF coupling constants (in Hz, red) derived from the cross-peak intensities. (a) Short-delay 1H,19F correlation experiment of GB1-1, recorded over 15 h. (b) Same as (a) but for GB1-2, recorded over 13 h. (c) Spectra of GB1-d. The left panel shows the 19F-detected [1H,1H]-TOCSY spectrum recorded over 19 h with a mixing time of 34 ms. The cross-peaks with Hβ resonances of the same residue are connected by purple lines. Stereospecific resonance assignments are indicated with subscripts, where a 1 or a 2 refers to the 19F spin attached to the γ1 or γ2-carbon, respectively. The right panel shows the short-delay 1H,19F correlation experiment recorded over 15 h. This spectrum was recorded after months of storage, when intact protein mass spectrometry indicated extensive heterogeneity due to partial proteolytic digestion of the flexible C-terminal peptide in our construct (Table S1 in the Supplement), including the His6 tag and the TEV cleavage site (Fig. S4 in the Supplement).
The 19F-NMR signals were readily linked to the assigned 1H-NMR spectra using short-delay 1H,19F correlation experiments (Fig. 5; Tan et al., 2024) and [1H,1H]-TOCSY spectra followed by an INEPT transfer to 19F (Fig. S2). These experiments were particularly important for GB1-d as they provided intra-residue links between 19F resonances (Fig. 5c).
As expected for the electron-withdrawing effect of fluorine, the 1H-NMR signals of the fluorinated residues were shifted low-field relative to the wild-type protein. The 1Hβ resonances of the singly fluorinated residues were shifted by about 0.3 ppm, and those of the diFVal residues were shifted by about 0.5 ppm, except for residue 54, which is exposed to ring currents from W45. The 1Hα signals were shifted low-field by about 0.25 and 0.45 ppm in the singly and doubly fluorinated residues, respectively.
The relative chemical shifts of the protons of the CH2F groups were highly conserved in all three samples (Fig. 5a–c), with their average chemical shifts increasing in the following order of residues: 54 < 39 < 29 < 21. The only exception was the γ1 protons of residue 54 in GB1-d, which were slightly low-field of the γ2 protons of residue 39 (Fig. 5c). The ordering of 1H chemical shifts is also conserved relative to the wild-type protein if the average of the chemical shifts of both methyl groups in each valine residue of the wild-type protein is used as the reference. Likewise, the chemical shifts of the Hβ atoms follow the ordering in the wild-type protein. This highlights the conservation of the amino acid side-chain conformations in the 3D structure of the protein. As expected for diastereotopic protons, the 1H chemical shifts within a CH2F group were often different from each other (Fig. S3 in the Supplement).
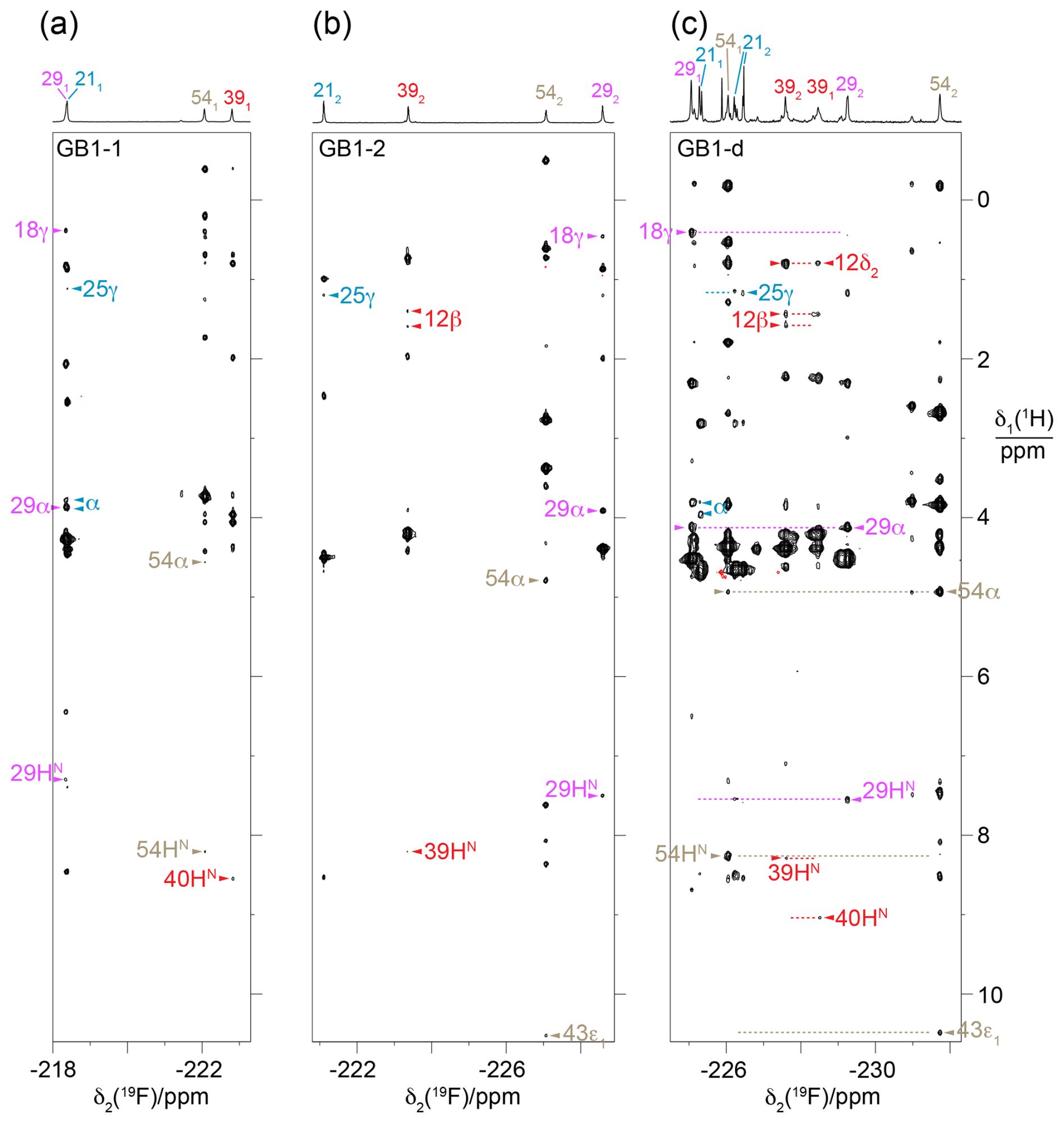
Figure 6HOESY spectra of GB1-1, GB1-2 and GB1-d recorded with a mixing time of 150 ms. The 1D 19F-NMR spectra are plotted above the 2D spectra and labelled with the 19F resonance assignments, indicating the stereospecific assignments with subscripts. The different fluorine signals and their cross-peaks are labelled with individual colours. Arrows identify assigned cross-peaks, with the 1H assignments indicated by the residue number and proton type. Two blue arrows labelled α identify NOEs with the CαH2 group of the glycine residue preceding M1 in the construct of GB1 used. (a) HOESY spectrum of GB1-1. Parameters used: t1max = 37.5 ms, t2max = 113 ms, total recording time of 36 h. (b) HOESY spectrum of GB1-2 recorded using the same parameters as in (a). (c) HOESY spectrum of GB1-d. The spectrum was recorded on a 500 MHz NMR spectrometer equipped with a 19F cryoprobe after the sample had aged, as evidenced by new signals and partial proteolytic digestion (Fig. S4). Parameters used: t1max = 18.3 ms, t2max = 133 ms, total recording time of 21 h. Dotted horizontal lines link to the locations of putative cross-peaks for the other CH2F group of the diFVal side chains.
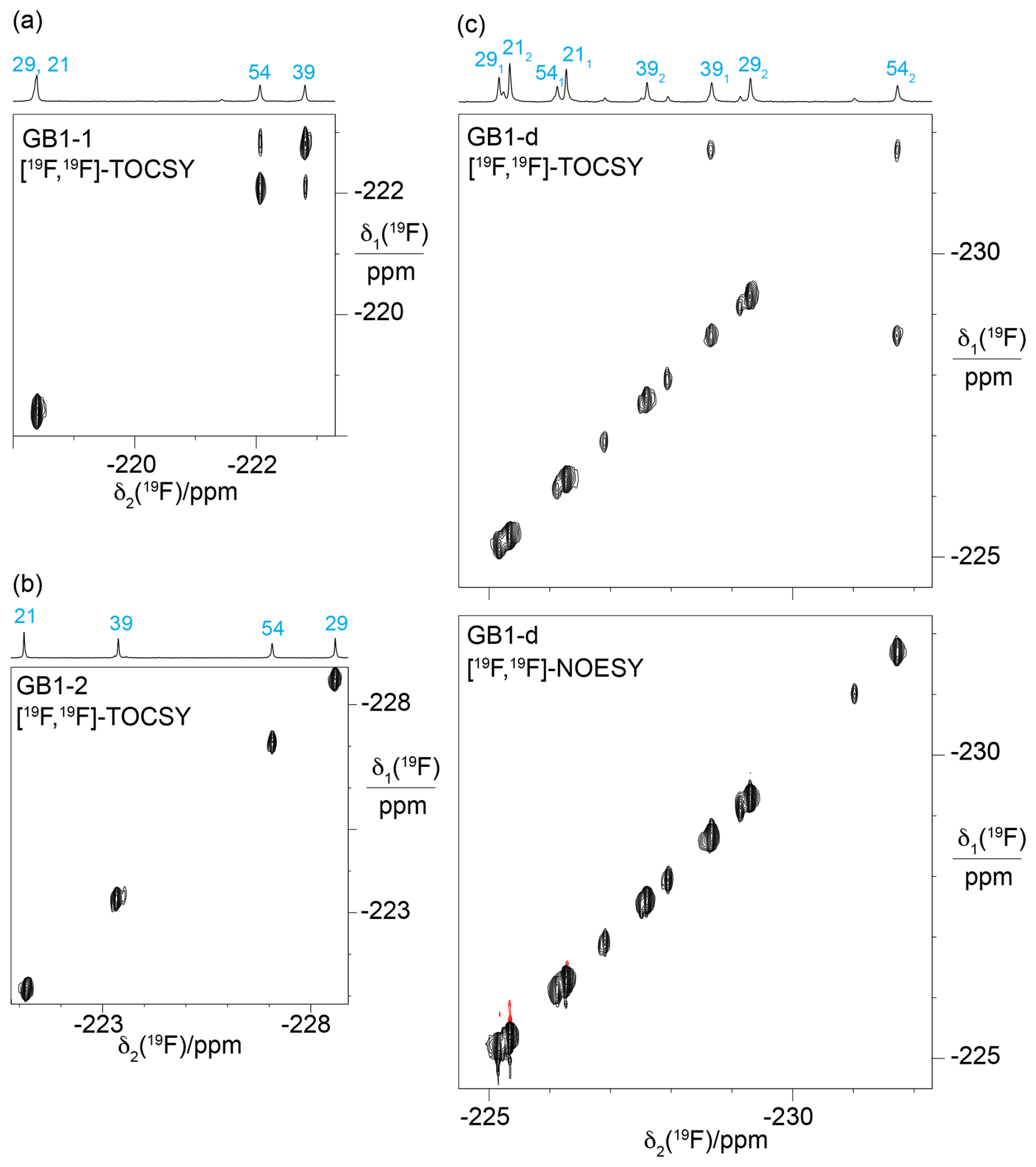
Figure 7[19F,19F]-TOCSY spectra showing through-space scalar 19F–19F couplings, TSJFF. Parameters used: 80 ms mixing time (DIPSI-2 mixing), t1max = 4.4–5.5 ms, t2max = 113 ms. The 1D 19F-NMR spectra with the resonance assignments are plotted above the spectra. (a) [19F,19F]-TOCSY spectrum of GB1-1 recorded over 5.5 h. (b) [19F,19F]-TOCSY spectrum of GB1-2 recorded in 2.1 h. (c) [19F,19F]-TOCSY spectrum of GB1-d recorded over 5.5 h (top panel) compared with the [19F,19F]-NOESY spectrum recorded in about 23 h with a mixing time of 150 ms (bottom panel).
2.4 Stereospecific 19F-NMR assignments in GB1-d
The stereospecific assignments of the 19F-NMR resonances of GB1-d were obtained by nuclear Overhauser effects (NOEs) observed in a [1H,19F]-HOESY spectrum. The strongest NOEs were observed for the buried residue 54, and the weakest were observed for the most highly solvent-exposed residue 21. The NOEs supported the conservation of the dihedral angles χ1, to be as in the wild-type protein. For example, the trans relationship between the Hα and Hβ atoms of residue 29 (Fig. 4b) is supported by intense intra-residual NOEs of both 19F spins with the Hα atom. In this geometry, the Cγ2H2F group is closer to the amide proton than the Cγ1H2F group, which is expected to show a long-range NOE with the Cγ2H3 group of T18. The HOESY spectrum confirms these NOEs (Fig. 6c). [1H,1H]-NOEs also assist. For example, the side-chain conformation of residue 39 (Fig. 4b) predicts an intra-residual [1H,1H]-NOE between the amide proton and the Cγ2H2F group but not the Cγ1H2F group. The assignment is confirmed further by an NOE of the γ2-fluorine with the Cδ2H3 group of L12 in the HOESY spectrum, whereas the γ1-fluorine makes an NOE with the amide proton of D40. The stereospecific assignments of residue 21 are less straightforward as its 3J(Hα,Hβ) coupling is compatible with two different staggered rotamers about the Cα–Cβ axis. Based on the side-chain conformation shown in Fig. 4b, which is reported both by the X-ray and NMR structures (Gallagher et al., 1994; Juszewski et al., 1999) and supported by the weak intensity of the intra-residual [1H,1H]-NOE between the 1Hβ and 1HN resonances, the stereospecific assignment of this residue was based on long-range NOEs between the γ2-fluorine and the methyl group of T25 and between the γ1-fluorine and the α protons of the glycine residue preceding M1 in our construct (Fig. 6). For residue 54, the HOESY spectrum unambiguously underpins the stereospecific assignments and the conformation of Fig. 4b by intra-residual NOEs between the γ1-fluorine and the amide proton and between the γ2-fluorine and the α-proton. In addition, the γ2-fluorine shows a well-resolved NOE with the ε1-proton of the side chain of W43 (Fig. 4c). The stereospecific 19F resonance assignments of GB1-d recapitulate the relative chemical shifts observed in GB1-1 and GB1-2, except for residues 21 and 39, which show the smallest chemical shift differences in GB1-d.
2.5 Through-space scalar 19F–19F couplings
[19F,19F]-TOCSY spectra offer a sensitive way of detecting through-space scalar 19F–19F couplings (TSJFF; Orton et al., 2021; Tan et al., 2025). In the 3D structures of GB1 (PDB ID 3GB1 and 1PGA; Juszewski et al., 1999; Gallagher et al., 1994), the Cγ1 atoms of V39 and V54 are within 4.1 Å of each other. Based on a C–F bond length of 1.40 Å and a van der Waals radius of fluorine of 1.47 Å, a direct 19F–19F contact between the FVal1 residues in GB1-1 is conceivable (Fig. 4a). Indeed, the [19F,19F]-TOCSY spectrum of GB1-1 showed the expected cross-peak, but its intensity is weak (Fig. 7a). As expected for the much greater distance between the Cγ2H2F groups (Fig. 4a), GB1-2 did not display a [19F,19F]-TOCSY cross-peak (Fig. 7b). Interestingly, the [19F,19F]-TOCSY spectrum of GB1-d showed no cross-peak between the γ1-fluorines, instead showing a cross-peak between the γ1-fluorine of residue 39 and the γ2-fluorine of residue 54 (Fig. 7c). While the solution structure 3GB1 reports the V39 Cγ1–V54 Cγ2 distance to be 4.4 Å, it is 3.8 Å in the crystal structure 1PGA. This contact thus suggests that the incorporation of diFVal residues favours the crystal structure conformation, where the dihedral angle χ1 of V54 is 50° versus 72° in the solution structure. It is well known that the fluorine atoms in 1,3-difluoropropane influence the rotamer populations of the CH2F groups. In particular, conformations with intramolecular contacts between the fluorine atoms are unfavourable (Lu et al., 2019; Marstokk and Møllendal, 1997; Wu et al., 1998), explaining the absence of intra-residual cross-peaks in the [19F,19F]-TOCSY spectrum of GB1-d (Fig. 7c).
The 19F–19F contacts reported by the TOCSY spectra must be short-lived, as no corresponding cross-peaks could be detected in [19F,19F]-NOESY spectra recorded with longer mixing time and measurement time. In addition, the absence of exchange cross-peaks in the [19F,19F]-NOESY spectrum of GB1-d (Fig. 7c) support the conclusion that the minor peaks observed in the 1D 19F-NMR spectrum stem from chemical rather than conformational heterogeneity.
To determine the size of the TSJFF couplings, we measured the relative intensity of the cross-peaks relative to the diagonal peaks in [19F,19F]-TOCSY spectra recorded with increasing mixing time. This peak ratio indicated that the TSJFF coupling is about 1.5 Hz in GB1-1 and 2.7 Hz in GB1-d (Fig. 8).
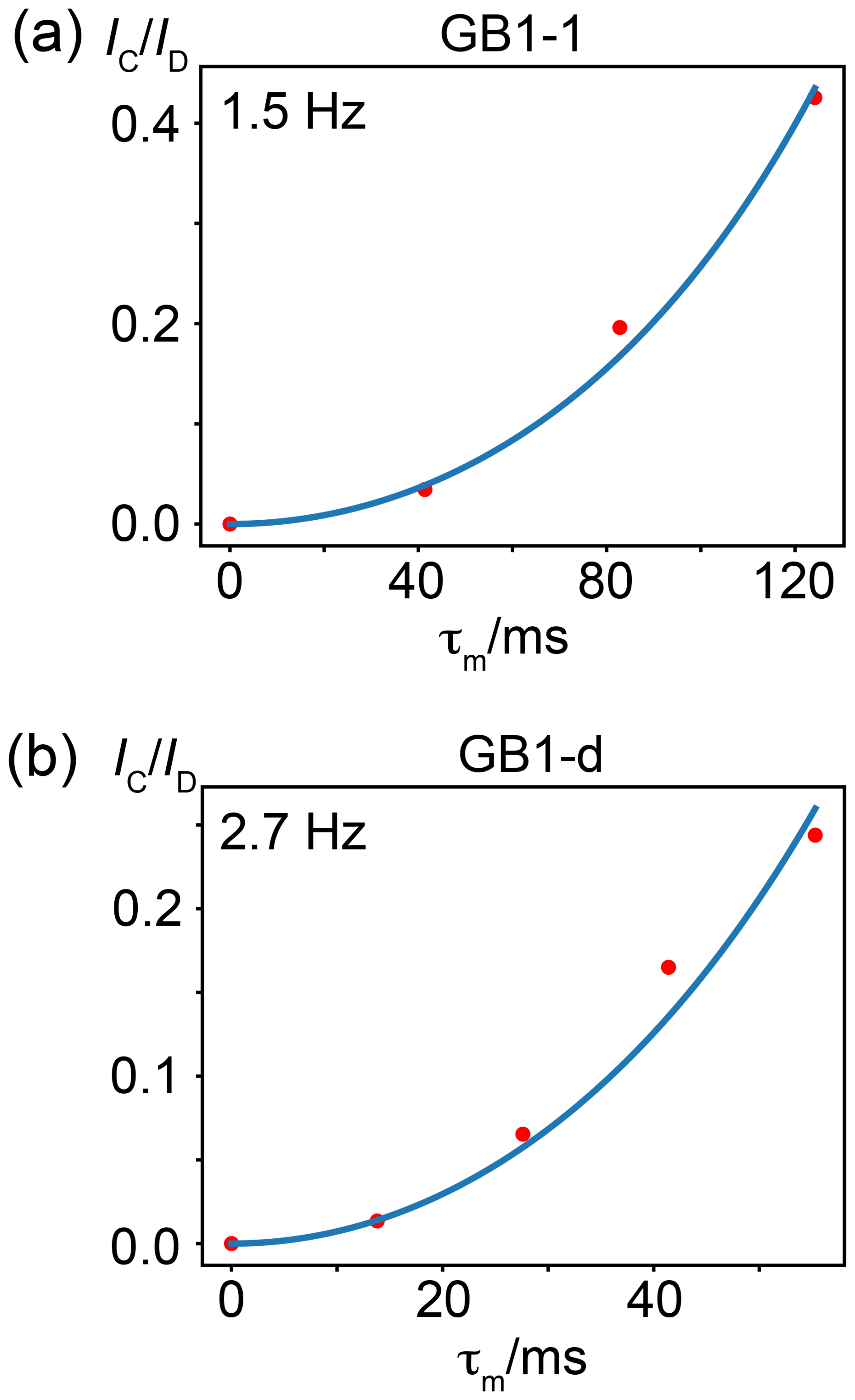
Figure 8Through-space 19F–19F couplings, TSJFF, in GB1-1 and GB1-d determined from [19F,19F]-TOCSY spectra recorded with increasing mixing time τm. The ratio of cross-peak (IC) over diagonal-peak (ID) intensity is plotted versus the TOCSY mixing time τm. The peak intensities were measured by integrating the respective peak intensities in 1D cross-sections. The blue curve shows the fit of the function = tan2(πJFFτm) (Braunschweiler and Ernst, 1983). The TSJFF coupling determined by a best fit (blue line) is indicated. (a) Coupling between the γ1-fluorine atoms of residues 39 and 54 in GB1-1. (b) Coupling between the γ1-fluorine of residue 39 and the γ2-fluorine of residue 54 in GB1-d. Data recorded at a 1H-NMR frequency of 500 MHz.
2.6 Rotamers of CH2F groups: restraints from 3JHF couplings
The 1.3 Å crystal structure of the protein PpiB produced with the uniform substitution of valine by FVal1 demonstrated a strong preference of the CH2F groups for staggered rotamers (PDB ID: 9C5D; Frkic et al., 2024b). The energy barrier between the three rotamers of the CH2F group in 1-fluoropropane was reported to exceed 4 kcal mol−1 (Feeney et al., 1996), highlighting the unfavourable nature of eclipsed conformations. Therefore, it is reasonable to describe the conformations of the CH2F groups in terms of staggered rotamers with different populations.
The Karplus curve for 3JHF couplings in aliphatic molecules indicates that the 19F spin couples with the Hβ atom with coupling constants of about 44 and 8 Hz for torsion angles of 180° (trans) and ± 60° (gauche), respectively (Williamson et al., 1968; Gopinathan and Narasimhan, 1971). Density functional theory (DFT) calculations performed for the 16 FVal1 residues in PpiB indicated somewhat different coupling constants, namely 30 ± 1 and 9 ± 3 Hz for the 3JHF couplings associated with the trans and gauche rotamers, respectively (Frkic et al., 2024b). The 3JHF coupling of 38 Hz measured for the γ2-fluorine in position 54 of GB1-d and GB1-2 thus identifies the rotamer that positions the fluorine atom trans relative to the 1Hβ atom (Fig. 9).
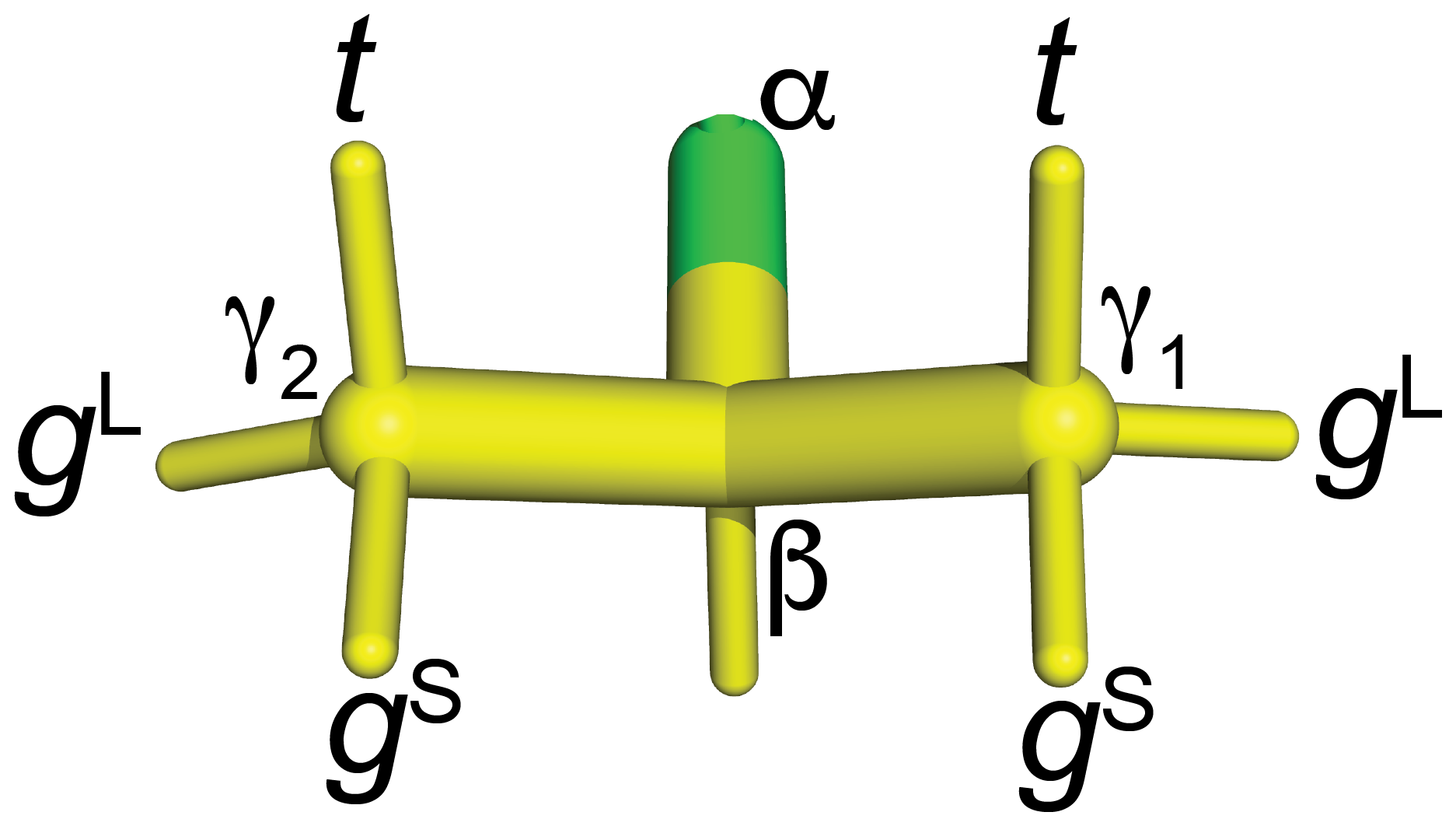
Figure 9Naming convention used in the present work to report the staggered rotamer conformations of CH2F groups in fluorinated valines. 3JHF is large when the fluorine atom is in the t position. In the gL rotamers, the fluorine atom is in the same plane as the Cγ1, Cβ and Cγ2 atoms. In this conformation of the CH2F group, a γ effect produces a relatively large up-field change in the 13C chemical shift of the CH3 group (by 6.3 ppm versus 5.1 ppm), and 3JHF between 1Hβ and 19F is small. For fluorine in the gS positions, the γ effect is relatively small and 3JHF is small.
2.7 Rotamers of CH2F groups: restraints from the γ effect on 13C chemical shifts
The 3JHF coupling discriminates between gauche and trans conformations but not between the two gauche rotamers. To distinguish between the gauche rotamers, we use the γ effect of 13C chemical shifts in X–C–C–13C moieties, where the non-hydrogen substituent X causes a pronounced up-field shift in the 13C resonance. Empirically, the effect is greatest for fluorine substitutions, which change the 13C chemical shift by about −6.8 ppm (Günther, 2013). The distinction between different rotamers relies on correction terms describing the dependence of the γ effect on the dihedral angle between the 13C and 19F spin. Conformational corrections of −1.0 and +2.0 ppm have been reported for the torsional angles of ± 60 and 180°, respectively (Fürst et al., 1990). Using the rotamer nomenclature of Fig. 9, the γ effect thus identifies the gL rotamer but does not distinguish between the gS and t rotamers. The rotamer information gained from the γ effect on 13C chemical shifts is therefore complementary to the information obtained from 3JHF couplings.
Figure 10 demonstrates how the γ effect manifests in large up-field changes in the 13C chemical shifts of the FVal methyl groups in the 13C-HSQC spectra of GB1-1 and GB1-2 compared with the wild-type protein. The γ effects range between about 5.8 and 7.4 ppm. In contrast, the 1H chemical shift changes in the FVal methyl groups were much smaller, down-field and quite uniform in size apart from the methyl group of residue 54 in GB1-2, which is governed by ring currents from W43. Even the methyl groups of residue 54 changed their 1H chemical shifts by less than 0.25 ppm.
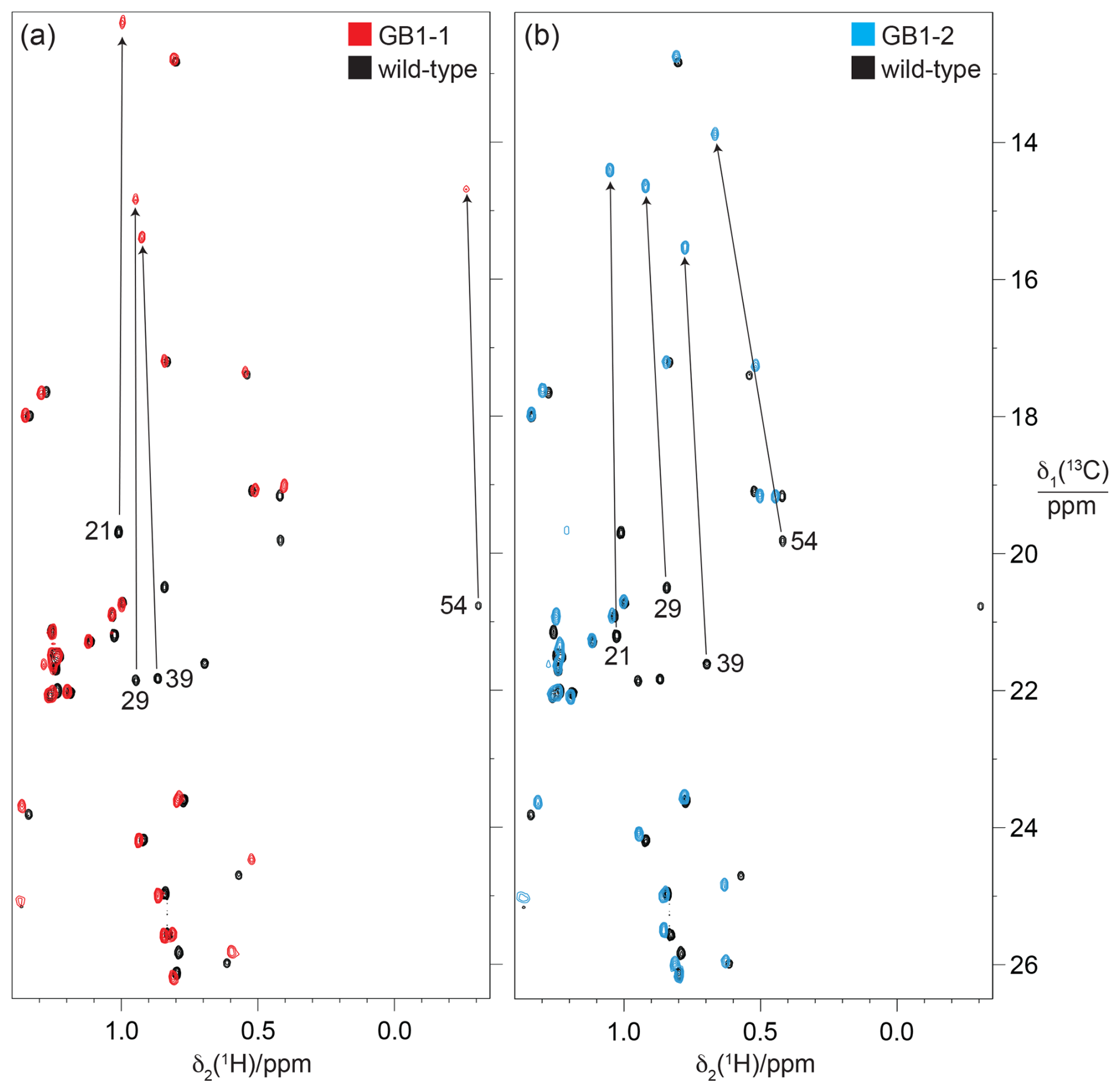
Figure 10Methyl region of the 13C-HSQC spectra of GB1-1 and GB1-2 superimposed onto the spectrum of wild-type GB1. The spectra were recorded at natural isotopic abundance using a 1H-NMR frequency of 800 MHz. (a) Spectrum GB1-1 (red) superimposed onto the spectrum of wild-type GB1 (black). The cross-peaks of the Cγ2H3 groups of valine residues in wild-type GB1 are labelled with the residue number. Arrows point to the corresponding cross-peaks in GB1-1. The stereospecific assignments in the wild-type protein were reported by Goehlert et al. (2004). (b) Same as (a) but for GB1-2 (blue spectrum), showing the change in chemical shifts for the valine Cγ1H3 groups.
In principle, the γ effects displayed by the 13C chemical shifts are modulated by the chemical environment, such as the presence or absence of fluorine atoms in nearby residues. Importantly, however, the 13C chemical shifts in GB1-1 and GB1-2 appear to respond little to the presence of nearby FVal residues, as shown by the methyl groups of alanine, leucine and threonine located within NOE distance of the fluorovaline side chains, which changed their 13C chemical shifts by less than 0.3 ppm (Fig. 10). This indicates that the conformational correction term of the γ effect easily exceeds the impact on 13C chemical shifts arising from changes in the local environments by FVal labelling.
2.8 Rotamers of CH2F groups: restraints from 3JFC coupling constants
If the magnitude of the γ effect is significantly determined by the rotamer states of the CH2F groups, it should correlate with the 3JFC couplings of the CH3 groups of the FVal residues. Therefore, we measured the 3JFC couplings of the CH3 groups of the FVal residues in GB1-1 and GB1-2. As the samples were at natural isotopic abundance, we used the constant-time 13C-HSQC difference experiment (Grzesiek et al., 1993) for best sensitivity. The coupling constants measured varied between 5.0 and 8.6 Hz. The largest coupling constants were measured for the FVal1 residues 21 and 29 in GB1-1 (8.6 and 8.3 Hz, respectively; Fig. 11). As expected, the up-field changes in the 13C chemical shifts of the FVal methyl groups, Δδ(13C), correlate with the 3JFC couplings (Fig. 12a).
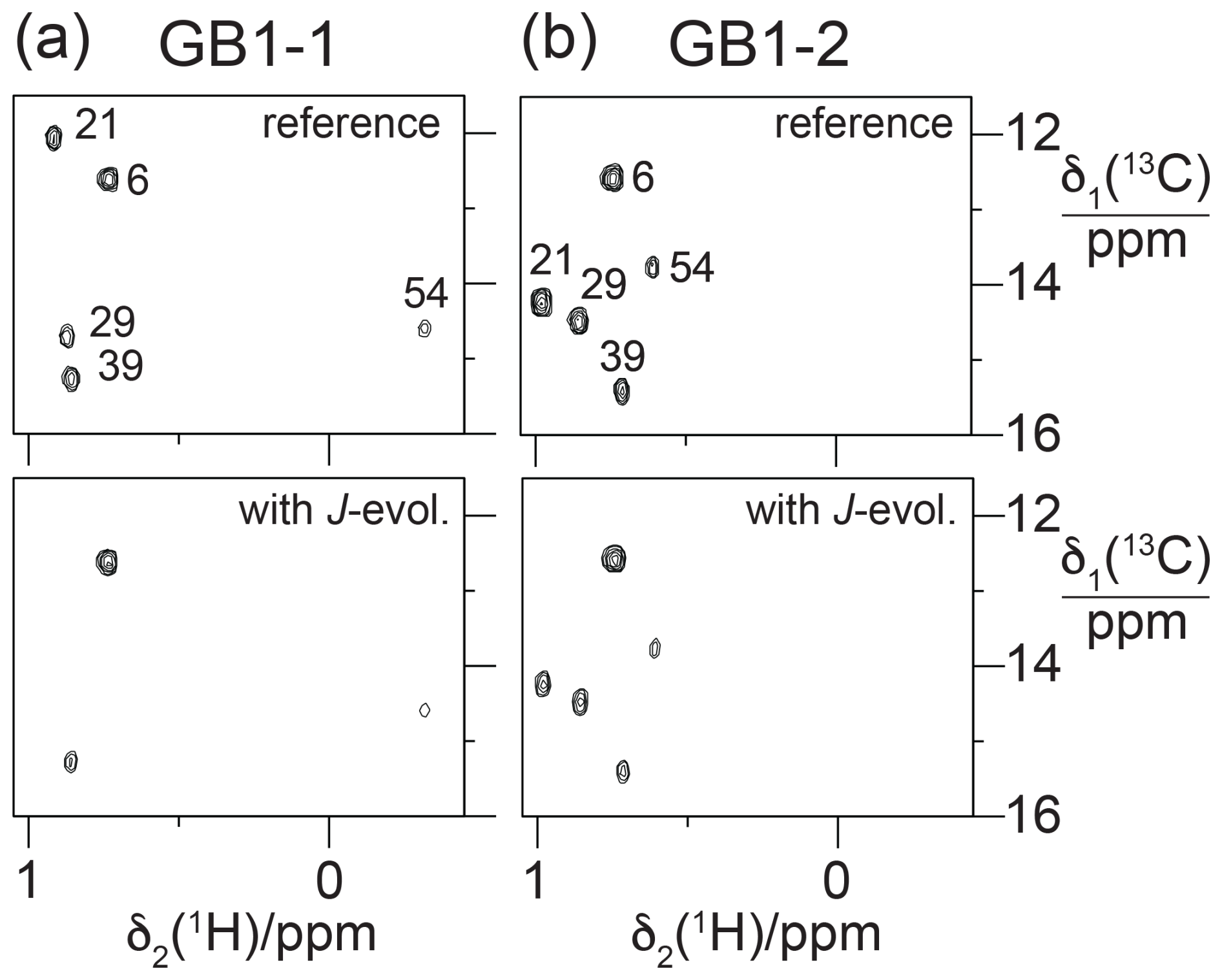
Figure 11Constant-time 13C-HSQC difference experiments for measuring 3JFC couplings between 19F and the 13C spin of the CH3 group (Grzesiek et al., 1993). Spectra with and without JFC coupling evolution were recorded in an interleaved manner. The experiment used the Bruker pulse program hsqcctetgpjclr with adaptation for JFC instead of JCC couplings. Parameters used: t1max = 19 ms, t2max = 82 ms, total recording time of 10 h. Spectra recorded on a 500 MHz NMR spectrometer equipped with a 1H/19F/13C cryoprobe. (a) Spectra obtained with GB1-1. The top panel shows the reference spectrum recorded with refocused JFC coupling, whereas JFC couplings evolved for a period of 57.5 ms in the spectrum shown in the bottom panel. The cross-peak intensities are conserved for Ile6, which does not couple to 19F. (b) Same as (a) but for GB1-2.
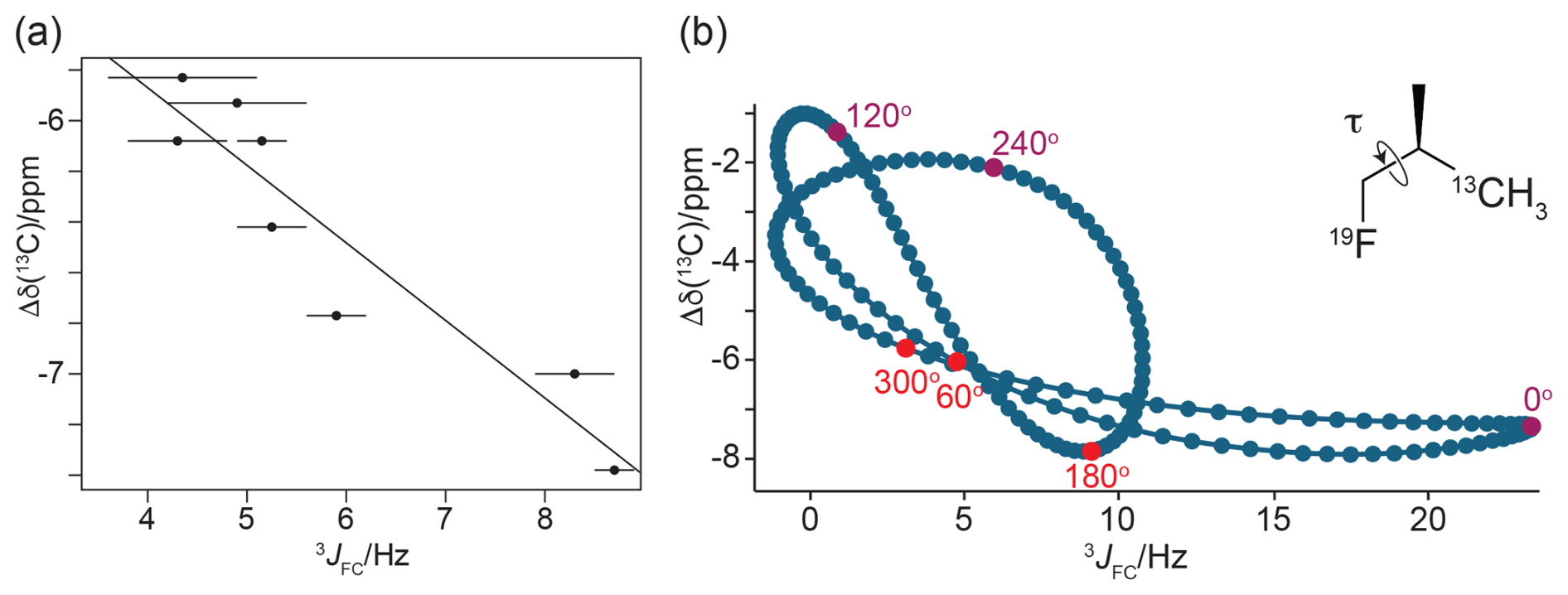
Figure 12γ effects of the 13C chemical shifts of FVal methyl groups generated by the fluorine atom in the CH2F group, Δδ(13C). (a) Correlation of Δδ(13C) with the 3JFC coupling constants. Δδ(13C) is calculated as the 13C chemical shift of FVal-labelled GB1 minus the 13C chemical shift of the corresponding valine residue in wild-type GB1. Error bars reflect the uncertainties in cross-peak intensities. The straight line (determined by a least-squares fit) was inserted to guide the eye. (b) Correlation of Δδ(13C) with the 3JFC coupling constants obtained by DFT calculations for (2R)-1-fluoro-2-methylpropane(3-13C), where only the torsion angle is varied and all other coordinates are fixed. The vertical axis reports Δδ(13C) relative to the methyl 13C chemical shift calculated for isobutane. The insert shows the chemical structure of the 1-fluoro-2-methylpropane compound. For the torsion angle τ = 0, the 19F and 13C nuclei are syn with respect to the intervening carbons and in the same plane. The plot highlights the points calculated for the torsion angles of the staggered conformations (red) and the energetically unfavourable eclipsed conformations (magenta). The pseudolinearity of the correlation between Δδ(13C) and 3JFC values obtained for the staggered conformations strengthens when all molecular degrees of freedom are relaxed in the DFT calculations (Fig. S8).
2.9 Density functional theory (DFT) calculations
For an independent underpinning of the γ effect, we performed DFT computations on FVal1 residues in PpiB, for which a high-resolution crystal structure is available with FVal1 residues at 16 sites (Frkic et al., 2024b). The calculations indicated that the presence of the γ1-fluorine atom changes the chemical shift of the 13Cγ2 spin by −6.3 ± 0.4 ppm if the intervening dihedral angle for rotation about the Cβ–Cγ1 bond is 169 ± 6° (i.e. the range of angles found following energy minimisation of the FVal1 side-chain conformations). In contrast, the 13Cγ2 chemical shift was calculated to change only by −5.1 ± 0.2 and −5.1 ± 0.3 ppm for the dihedral angles −61 ± 6 and 60 ± 2°, respectively.
To explore the full range of the γ effect independently of the protein context, DFT calculations were performed using the model compound 1-fluoro-2-methylpropane with a systematic change in the torsion angle τ (Fig. 12b). Two sets of torsion-dependent calculations were performed to evaluate the sensitivity of the DFT calculations to minor changes in bond lengths and bond angles: one where only the relevant torsion angle was varied and where all other atomic positions were fixed (Figs. 12b and S7 in the Supplement) and a second one where all other degrees of freedom were relaxed for each specified torsion angle (Fig. S8 in the Supplement). Both sets of calculations confirmed that the γ effect is significantly larger for the torsion angle τ = 180° than for τ = ± 60° (Figs. 12b, S7 and S8). The main difference in the two sets of calculations is that the 3JFC coupling constant for τ = 0° was smaller with optimisation of the other molecular degrees of freedom (Figs. S7 and S8).
2.10 Rotamers determined by 3JHF couplings, 3JFC couplings and the γ effect on 13C chemical shifts
According to the Karplus curve, the FVal residues possess larger 3JFC constants for the gL than the gS and t rotamers. In rigid organic molecules containing C–C–C–F moieties, 3JFC values of about 10 Hz were reported for dihedral angles of 180° (Barfield et al., 1978). The smaller 3JFC constants measured for GB1-1 and GB1-2 thus suggest that the CH2F groups of the FVal residues are subject to some degree of motional averaging. Nonetheless, our data show evidence of preferential rotamer populations.
Table 1Determination of CH2F rotamers from 3JHF couplings, 19F chemical shifts, 13C chemical shift changes and 3JFC coupling constants a.
a The 3JHF couplings were measured by line fitting in 1D 19F-NMR spectra and short-delay 1H,19F correlation experiments (Tan et al., 2024). For the Cγ2H2F group of residue 54, the 3JHF coupling was measured from the peak splittings observed for the Cγ2H2–Hβ NOESY cross-peaks. b Where two rotamers are listed, the first rotamer appears to be more highly populated than the second rotamer, which is more highly populated than the third rotamer. In the case of residue 39 in GB1-1, the data suggest a preference for the gS rotamer, while a preference of the gL versus t rotamer is difficult to discern. c The data for GB1-1 pertain to the Cγ2H3 group, and the data for GB1-2 pertain to the Cγ1H3 group. The γ effect on 13C chemical shifts, Δδ(13C), is reported as δ(13C) of the FVal-labelled protein minus the δ(13C) value of the wild-type protein. d Coupling constant of 19F to the carbon of the CH3 group measured in a spin-echo difference experiment (Fig. 11). e Based on 3JHF, Δδ(13C) and 3JFC. See Fig. 9 for the nomenclature used.
For example, the Cγ2H2F group of residue 54 in GB1-2 and GB1-d clearly populates the t rotamer, as evidenced by the large 3JHF coupling and up-field 19F chemical shifts. As expected for this conformation, the γ effect of the 13Cγ1 spin in GB1-2 is relatively small (−5.93 ppm versus the average of −6.4 ppm; Table 1; Fig. 10b). In addition, the 3JFC coupling of the Cγ2H2F group of residue 54 in GB1-2 is relatively small, as expected for a dihedral angle of about −60° (5 Hz; Table 1).
The data are less decisive with regard to the rotamer preference of the Cγ1H2F group of residue 54. In GB1-1, its 3JHF coupling is relatively small, suggesting a g rotamer, but it is not as small as in GB1-d (Table 1). Its 19F chemical shift is low-field but not as much as for other residues, suggesting an admixture of the t rotamer. The γ effect is relatively small but not as small as expected if only gS or t rotamers were populated. However, the very small 3JFC coupling in GB1-1 (4.3 Hz) excludes a significant population of the gL rotamer. In summary, the gS rotamer appears to be preferred. A better fit would have to consider additional rotamers and deviations from the perfectly staggered conformations.
Similarly, the rotamer analysis of other CH2F groups does not indicate single rotamers, but preferential populations can be identified. For example, the largest γ effects of FVal residues were observed for the 13Cγ2 spins of residues 21 (−7.38 ppm) and 29 (−7.00 ppm) in GB1-1. These shifts, as well as relatively large 3JFC couplings, suggest that the respective FVal1 residues preferentially populate the gL rotamer. As expected for this rotamer, the 3JHF couplings of both FVal1 residues are small, and their 19F chemical shifts are down-field (Fig. 2a).
Table 1 and Fig. 13 summarise the preferential rotamers for the FVal residues in GB1-1 and GB1-2, derived from 3JHF couplings, 3JFC couplings and the γ effect of the 13C chemical shift of CγH3 groups with the assumption of staggered rotamers.
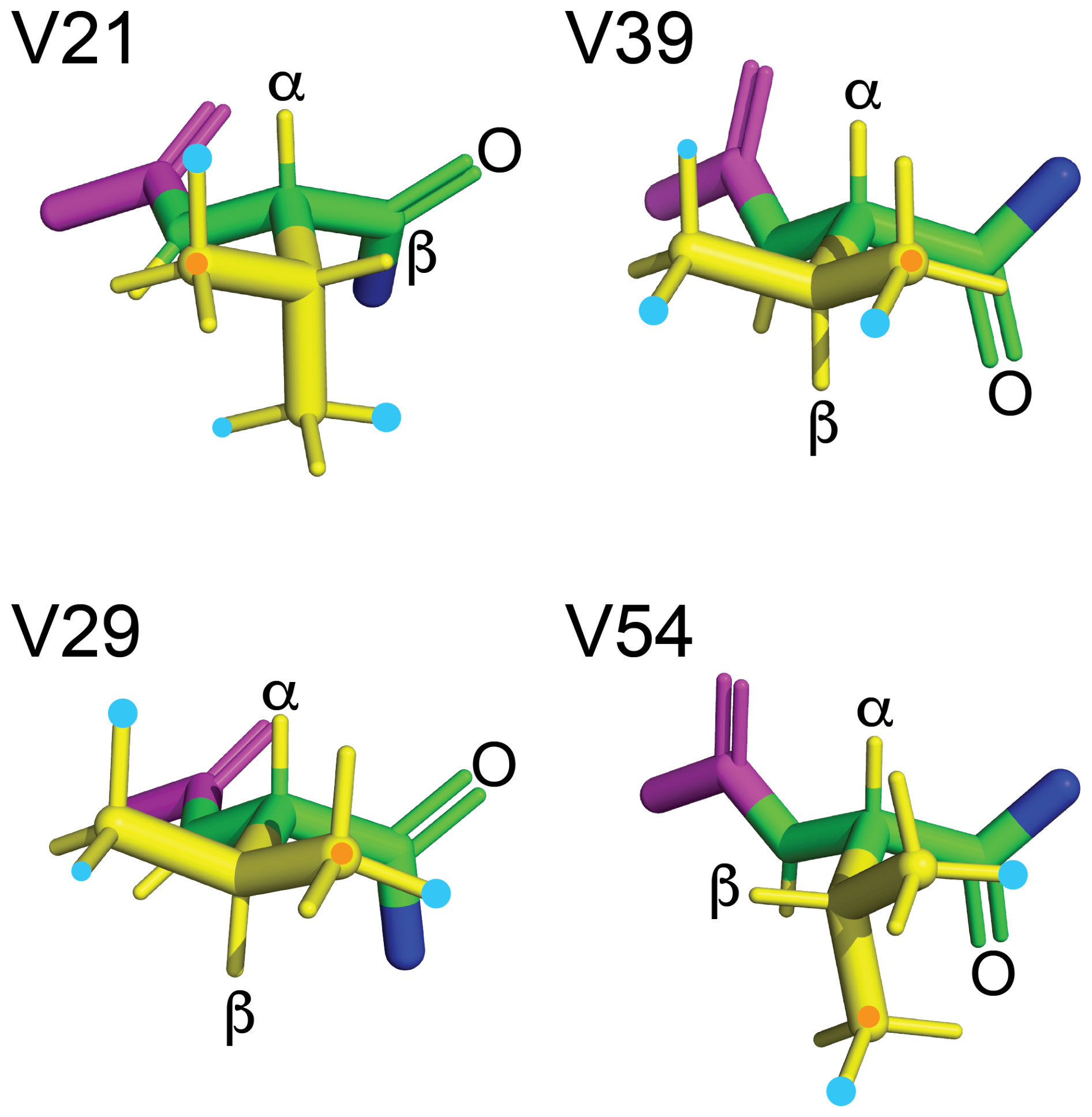
Figure 13Rotamers preferentially populated by fluorovaline residues in GB1-1 and GB1-2. The four valine side chains are shown in the conformation of the solution structure 3GB1 with the methyl groups rotated into fully staggered conformations. Cyan dots mark the preferred locations of fluorine atoms in GB1-1 and GB1-2 based on the data of Table 1. When the data clearly point to the population of more than a single rotamer, the most highly populated rotamer is identified by a larger dot. An orange dot marks the γ1-carbon. The backbone atoms N, Cα, C′ and O are shown in green; the HN, Hα and side-chain atoms are shown in yellow; the Cα, C′ and O of the previous residue are shown in magenta; and the nitrogen of the following residue is shown in blue. The double bond of each carbonyl group is shown by two lines, and the carbonyl oxygen of each valine residue is labelled.
Assigning preferential rotamers in GB1-d is more difficult as the diFVal residues contain no slowly relaxing CH3 group, which makes 3JFC coupling measurements difficult. Furthermore, the 13C chemical shifts of the CH2F groups are under the influence of the directly bonded fluorine atoms. While the γ effect on 13C chemical shifts should also be sensed by the α-carbons, a qualitative analysis of GB1-1 showed that the 13Cα chemical shifts of the FVal residues varied much more than expected for the γ effect. Reliable restraints were therefore limited to the 3JHF couplings. For residue 54, the 3JHF couplings suggest that the diFVal side chain preferentially populates the same rotamers as the FVal1 and FVal2 analogues, except for the fact that the smaller 3JHF coupling of the γ1-fluorine suggests a greater preference for the gS rotamer.
In contrast, diFVal in position 21 showed a larger 3JHF coupling for the γ1-fluorine than GB1-1 (Table 1), suggesting a change in preferred rotamer to the t rotamer. Notably, a diFVal residue maintaining the gL rotamers indicated by the FVal1 and FVal2 data would lead to an all-trans conformation of the F–Cγ1–Cβ–Cγ2–F moiety, which has been shown to be energetically unfavourable in 1,3-difluoropropane (Marstokk and Møllendal, 1997; Wu et al., 1998; Lu et al., 2019). Likewise, maintaining the gS rotamers identified for residue 39 as most highly populated in GB1-1 and GB1-2 would bring the two fluorines into close proximity in GB1-d, which is energetically unfavourable. In this case, the γ2-fluorine changes its rotamer preference for GB1-d in favour of the t rotamer, as indicated by an increased 3JHF coupling constant (Table 1). Despite the larger 3JHF coupling, however, the 19F-NMR signal of the γ2-fluorine is down-field of the γ1-fluorine signal in GB1-d, in apparent violation of the γ-gauche effect. Notably, however, the chemical environment of the γ1-fluorine changes in GB1-d, as evidenced by the switch in TSJFF coupling from the γ1-fluorine of residue 54 to the γ2-fluorine (Fig. 7a and c), compromising the interpretation of the 19F chemical shifts.
The rotamer assignments of the Cγ1H2F groups of residues 39 and 54 agree with the TSJFF coupling observed in the [19F,19F]-TOCSY spectrum of GB1-1 (Fig. 7a) as the NMR structure 3GB1 of the wild-type protein suggests that the shortest possible 19F–19F distance is between the γ1-fluorines of these residues if both populate the gS rotamer. These rotamers are, indeed, highly populated (Table 1). In contrast to the structure 3GB1, the crystal structures 1PGA, 1PGB (Gallagher et al., 1994) and 2QMT (Frericks Schmidt et al., 2007) show the γ1-carbon of Val39 to be closer to the γ2-carbon of Val54 (3.8 Å versus 4.2 Å). The additional fluorine atoms in GB1-d thus seem to drive the small structural change towards the conformation observed in the single crystal (Fig. 7c).
2.11 Fluorines near carbonyl carbons
The rotamer analysis shows that the FVal2 residues in positions 21 and 54 and the FVal1 residue in position 29 preferentially populate a rotamer that positions the fluorine atom in close proximity of the carbonyl carbon of the same residue (Fig. 13). This conformation, which results in a C–F distance as short as 2.85 Å, has been noted previously in the crystal structure of PpiB made with FVal1 (Frkic et al., 2024b). It suggests a favourable interaction between the negatively polarised fluorine and the positively polarised carbonyl carbon.
To explore whether these close fluorine–carbonyl carbon interactions are manifested in the 13C chemical shifts of the carbonyl carbons, we recorded HNCO experiments of GB1-1, GB1-2 and wild-type GB1 at natural 13C abundance. The wild-type protein was uniformly labelled with 15N by conventional expression in vivo, while the samples containing FVal1 or FVal2 were produced by cell-free protein synthesis from amino acids, where the amino acids following the valine sites in the amino acid sequence (asparagine, aspartic acid, phenylalanine and threonine) were labelled with 15N. This selective labelling approach allowed the resonance assignment from the first 2D plane of the HNCO experiment correlating 13C chemical shifts with backbone amide protons (Fig. S5 in the Supplement).
Most notably, the GB1-1 and GB1-2 samples showed significant up-field changes in the 13C chemical shifts of the carbonyl groups of the FVal1 and FVal2 residues, with GB1-2 showing the largest 13C chemical shift changes. The effect does not correlate with the distance between 19F and 13C spins, however, as large internuclear distances still produced large chemical shift changes as shown, for example, by residues 29 and 39 in GB1-2. In contrast, residue 54 showed, by far, the smallest change in the 13C chemical shift. Therefore, the changes in 13C chemical shifts are not a simple function of spatial proximity.
To explore whether fluorine changes chemical shifts mostly via through-bond or through-space effects, we recorded 15N-HSQC spectra (Fig. S6 in the Supplement). The amide cross-peaks of all fluorinated valine analogues shifted significantly, with all amide protons shifting down-field, while the amide nitrogens shifted up-field, except for residue 21 in GB1-2. The shifts observed for the diFVal residues in GB1-d corresponded, by and large, to the sum of the chemical shift changes observed for GB1-1 and GB1-2. This was also observed for the most strongly shifting cross-peaks of non-valine residues. This cumulative effect of the fluorination appears to be insensitive to the rotamer populations of the CH2F groups. Some of the largest 1H chemical shift changes were observed for the amide protons of residues following the valine sites. Comparing the chemical shift changes of residues 29 and 39, which feature the same torsion angle χ1 between the α- and β-carbons (Fig. 13), it is interesting to note their similarity in terms of the chemical shift changes of their own amide cross-peaks, as well as the amide cross-peaks of the following residues. As through-bond effects, these effects would be active six bonds from the 19F spin, but the conservation of the χ1 angle could also entail a conserved structural perturbation.
3.1 Effects of fluorinated valines on protein fold stability
Highly conserved 1H chemical shifts confirm that the 3D structure of GB1 is fully maintained when all four valine residues are replaced by fluorinated valine analogues, but their presence destabilises GB1 with respect to heat denaturation. Owing to the shorter side chain, the CH2F groups of the fluorinated valine analogues are closer to the backbone than those of correspondingly fluorinated leucine analogues, leaving the CH2F groups with fewer options to be accommodated in the 3D protein fold. Nonetheless, the melting temperatures of the GB1 variants made with fluorinated valines did not decrease much more than for GB1 samples made with fluorinated leucine (Tan et al., 2025). We previously noted that the substitution of valine for FVal1 in the protein PpiB, which contains 16 valine residues, reduced the melting temperature by only 11 °C (Frkic et al., 2024b), i.e. only a little more than the substitution of five leucine residues by fluorinated analogues (Tan et al., 2024).
The rotamers preferred by the CH2F groups of FVal1 and FVal2 residues often position the fluorine atom next to the carbon of backbone carbonyl groups (Fig. 13) despite the nominal van der Waals radii suggesting steric hindrance. Examples of this conformation have been observed previously in the high-resolution crystal structure of FVal1-labelled PpiB (Frkic et al., 2024b). It has been argued that the standard van der Waals radii are not appropriate for sp2 carbons bound to electron-withdrawing moieties (Kruse et al., 2020). Oxygen atoms have been reported to form weakly stabilising bonds with carbonyl carbons (Bartlett et al., 2010) or simply engage in a favourable dipole–dipole interaction (Worley et al., 2012). The experimental results of the present work indicate that the interaction between fluorine and a carbonyl carbon may promote rotamers with short fluorine–carbon distances, but it is insufficient to lock the CH2F group into a single rotamer. The interaction does not manifest in unusually large changes in 13C chemical shifts of the carbonyl groups.
3.2 Preferential rotamer populations of CH2F groups
Each CH2F group can access three different staggered rotamers, which are separated by energy barriers of about 4–5 kcal mol−1 (Feeney et al., 1996). As shown by the 1.3 Å crystal structure of FVal1-labelled PpiB, deviations from perfectly staggered conformations are small, but the protein environment rarely confines the fluorine atoms to single rotamers (Frkic et al., 2024b). The present work confirms the population of multiple rotamers in GB1 for both solvent-exposed and buried CH2F groups. A strong preference for single rotamers was detected only for residue 54, which is the most deeply buried valine residue in wild-type GB1. Nonetheless, rotamer preferences can also be discerned for highly solvent-exposed CH2F groups, such as in residue 21. Interestingly, the HCl salts of the free amino acids dissolved in methanol already show evidence of preferential rotamer populations as the 3JHF and 3JFC couplings and 19F chemical shifts differ between the FVal1 and FVal2 amino acids. The values reported for FVal1 and FVal2, respectively, are 20.5 and 15.5 Hz for 3JHF, −224.55 and −222.17 ppm for δ(19F), and 7.5 and 7.3 Hz for the 3JFC coupling of the CH3 group (Maleckis et al., 2022). Most notably, the 3JHF couplings and 19F chemical shifts correlate as expected for the γ-gauche effect (Tonelli et al., 1982; Feeney et al., 1996; Tan et al., 2024). The correlation also holds for diFVal, where the 3JHF couplings are 20.8 and 23.6 Hz for the 19F-NMR signals at −223.93 and −225.72 ppm, respectively (Maleckis et al., 2022).
3.3 Through-space JFF couplings
As in previous work with fluorinated amino acids (Kimber et al., 1978; Orton et al., 2021; Tan et al., 2025), TSJFF couplings between fluorinated valine analogues were readily observed by [19F,19F]-TOCSY experiments. The couplings are much smaller than the TSJFF coupling of 21 Hz reported for 6-fluorotryptophan-labelled dihydrofolate reductase, where the fluorine atoms are held in a rigid structure relative to each other (Kimber et al., 1978). Therefore, the TSJFF couplings detected in the present work are evidence of direct contacts but not of attractive forces between the fluorine atoms despite the generally hydrophobic nature of carbon-bonded fluorine. To the contrary, the polarity of the C–F bonds may well discourage direct 19F–19F contacts, just as bond polarities restrict the accessible rotamer combinations in diFVal residues, hindering the detection of intra-residual [19F,19F]-TOCSY cross-peaks between the CH2F groups of diFVal.
3.4 γ effect on 13C chemical shifts
The γ effect and its dependence on the torsion angle of the intervening atoms have been known for a long time (Tonelli and Schilling, 1981; Fürst et al., 1990). A γ effect has also been reported for the 13C chemical shift of δ-methyl groups of leucine residues, where the Cα–Cβ and Cγ–Cδ bonds form a dihedral angle of 180° about the Cβ–Cγ bond (MacKenzie et al., 1996). 19F spins produce the largest γ effects (Günther, 2013). To the best of our knowledge, the present work is the first experimental demonstration of the dihedral angle dependence of the 13C-detected γ effect driven by CH2F groups. The γ effect also prevails for singly fluorinated leucine analogues in GB1, where the smallest Δδ(13C) values have been detected for the methyl groups of the buried residue 5, which is characterised by pronounced rotamer preferences of the CH2F groups (Tan et al., 2025). As the difference between the 13C chemical shifts of canonical valine residues and their fluorinated analogues, Δδ(13C), is also influenced by any structural adjustments caused by the fluorine atoms, the γ effect is a less straightforward parameter for interrogating dihedral angles than 3JFC couplings.
3.5 Using the γ effect in site-specific probes
As demonstrated by the current results, the hydrogen-fluorine substitution in a methyl group does not fully stop the rotation of the resulting CH2F group, even if it is buried in the core of a structurally stable protein. This highlights the ease with which fluorine atoms can be accommodated in the protein structure. As the γ effect translates changes in the population of different rotamers into a significant change in the 13C chemical shift of the remaining CH3 group, a fluorinated isopropyl probe could be used as a sensor of ligand binding, which can be interrogated with a spectrometer that is not equipped for 19F-NMR.
3.6 Sequence-specific assignment of 19F-NMR resonances
As chemical shift anisotropy adds a prominent relaxation mechanism for 19F spins, the broad 19F-NMR signals encountered for large proteins often call for resonance assignment by site-directed mutagenesis. Fortunately, much information can be drawn from the short-delay 1H,19F correlation experiment, which provided excellent sensitivity for linking 19F and 1H chemical shifts, even in the 19 kDa protein PpiB containing fluorinated leucine residues (Tan et al., 2024). The present study shows that different fluorinated valine residues in GB1 maintain the same relative order of average 1H chemical shifts in the CH2F groups as the valine CH3 groups in the wild-type protein. The same applies for the Hβ atoms that are also detected by the short-delay 1H,19F correlation experiment. Inspection of the NMR spectra reported previously for GB1 (Tan et al., 2025) and PpiB (Tan et al., 2024) containing fluorinated leucine residues likewise reveals full conservation of the relative ordering of the 1H chemical shifts of the Hγ atoms, as in the wild-type protein. In GB1, even the 1H chemical shifts of the CδH2F groups follow the average chemical shift ordering of the methyl groups in the wild-type protein. This greatly aids the 19F resonance assignments if the 1H assignments of the wild-type protein are available. Alternatively, genetic encoding of amino acids differing from their parents by a single H-to-F substitution has recently become possible in vivo (Qianzhu et al., 2022; 2024; 2025) and may also become possible for FVal and FLeu residues.
3.7 Comparison of the NMR properties of fluorinated valines and fluorinated leucines
In previous work, we substituted leucines for singly and doubly fluorinated analogues in GB1 (Tan et al., 2025) and PpiB (Tan et al., 2024). The leucine analogues are very similar to FVal1, FVal2 and diFVal, except for the presence of an additional CH2 group between the α-carbon and the isopropyl group, which adds flexibility to the amino acid side chains. As expected for increased side-chain mobility, the transverse 19F relaxation rates of diFLeu residues measured in GB1 were slower than for the diFVal-labelled sample. Furthermore, some of the 19F-NMR signals of PpiB made with FVal1 were very broad, suggesting exchange broadening. In contrast, none of the FLeu1 or FLeu2 signals showed similarly severe line broadening effects, and more uniform peak heights were obtained with the singly fluorinated leucine variants than with diFLeu, confirming the importance of local side-chain mobility for narrow 19F-NMR signals. In all examples, the signal heights were lowest for the most deeply buried CH2F groups.
Proteins prepared with either FVal1, FVal2, FLeu1 or FLeu2 produce 19F-NMR spectra with extraordinary spectral widths and are only slightly destabilised compared with their wild-type parents. The recent commercial availability of these amino acids greatly advances the accessibility of the corresponding proteins. The conservation of relative 1H chemical shifts associated with the conservation of the side-chain conformations opens a convenient route to assign the 19F-NMR spectra. The pronounced sensitivity of CH2F groups to changing protein environments renders them excellent NMR probes of, for example, ligand binding.
The NMR data are available at https://doi.org/10.5281/zenodo.17082808 (Otting, 2025).
The supplement related to this article is available online at https://doi.org/10.5194/mr-6-257-2025-supplement.
EHA prepared the protein samples and performed the 1D NMR measurements. NFC performed the DFT calculations. AM synthesised fluorinated valine analogues. GO coordinated the project, performed the 2D NMR measurements, and prepared the final paper and figures.
At least one of the (co-)authors is a member of the editorial board of Magnetic Resonance. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
We thank Roger Amos, Rika Kobayashi and Michael Collins for the preliminary DFT calculations of 19F chemical shifts and Martin Scanlon for the access to the 500 MHz NMR spectrometer equipped with a 1H/19F/13C cryoprobe.
This research has been supported by the Australian Research Council (ARC, grant no. DP230100079) and the ARC Centre of Excellence for Innovations in Peptide and Protein Science (grant no. CE200100012).
This paper was edited by Ad Bax and reviewed by Geoffrey Bodenhausen and two anonymous referees.
Barfield, M., Marshall, J. L., Canada, E. D., and Willcott III, M. R.: γ-methyl substituent effect on vicinal coupling constants involving carbon-13, J. Am. Chem. Soc., 100, 7075–7077, https://doi.org/10.1021/ja00490a056, 1978.
Bartlett, G. J., Choudhary, A., Raines, R. T., and Woolfson, D. N.: interactions in proteins, Nat. Chem. Biol., 6, 615–620, https://doi.org/10.1038/nchembio.406, 2010.
Braunschweiler, L. and Ernst, R. R.: Coherence transfer by isotropic mixing – application to proton correlation spectroscopy, J. Magn. Reson., 53, 512–528, https://doi.org/10.1016/0022-2364(83)90226-3, 1983.
Feeney, J., McCormick, J. E., Bauer, C. J., Birdsall, B., Moody, C. M., Starkmann, B. A., Young, D. W., Francis, P., Havlin, R. H., Arnold, W. D., and Oldfield, E.: 19F nuclear magnetic resonance chemical shifts of fluorine containing aliphatic amino acids in proteins: studies on Lactobacillus casei dihydrofolate reductase containing (2S,4S)-5-fluoroleucine, J. Am. Chem. Soc., 118, 8700–8706, https://doi.org/10.1021/ja960465i, 1996.
Frericks Schmidt, H. L., Sperling, L. J., Gui Gao, Y., Wylie, B. J., Boetcher, J. M., Wilson, S. R., and Rienstra, C. M.: Crystal polymorphism of protein GB1 examined by solid-state NMR spectroscopy and X-ray diffractions, J. Phys. Chem. B, 111, 14362–14369, https://doi.org/10.1021/jp075531p, 2007.
Frkic, R. L., Tan, Y. J., Abdelkader, E. H., Maleckis, A., Tarcoveanu, E., Nitsche, C., Otting, G., and Jackson, C. J.: Conformational preferences of the non-canonical amino acids (2S,4S)-5-fluoroleucine, (2S,4R)-5-fluoroleucine, and 5,5'-difluoroleucine in a protein, Biochemistry, 63, 1388–1394, https://doi.org/10.1021/acs.biochem.4c00081, 2024a.
Frkic, R., Tan, Y. J., Maleckis, A., Otting, G., and Jackson, C. J.: 1.3 Å crystal structure of E. coli peptidyl–prolyl isomerase B with uniform substitution of valine by (2S,3S)-4-fluorovaline reveals structure conservation and multiple staggered rotamers of CH2F groups, Biochemistry, 63, 2602–2608, https://doi.org/10.1021/acs.biochem.4c00345, 2024b.
Fürst, A., Pretsch, E., and Robien, W.: Comprehensive parameter set for the prediction of the 13C-NMR chemical shifts of sp3-hybridized carbon atoms in organic compounds, Anal. Chim. Acta, 233, 213–222, https://doi.org/10.1016/S0003-2670(00)83481-9, 1990.
Gallagher, T., Alexander, P., Bryan, P., and Gilliland, G. L.: Two crystal structures of the B1 immunoglobulin-binding domain of streptococcal protein G and comparison with NMR, Biochemistry, 33, 4721–4729, https://doi.org/10.1021/bi00181a032, 1994.
Goehlert, V. A., Krupinska, E., Regan, L., and Stone, M. J.: Analysis of side chain mobility among protein G B1 domain mutants with widely varying stabilities, Prot. Sci., 13, 3322–3330, https://doi.org/10.1110/ps.04926604, 2004.
Gopinathan, M. S. and Narasimhan, P. T.: Finite perturbation molecular orbital approach to the dihedral angle dependence of vicinal H-H and H-F couplings, Mol. Phys., 21, 1141–1144, https://doi.org/10.1080/00268977100102271, 1971.
Grzesiek, S., Vuister, G. W., and Bax, A.: A simple and sensitive experiment for measurement of JCC gouplings between backbone carbonyl and methyl carbons in isotopically enriched proteins, J. Biomol. NMR, 3, 487–493, https://doi.org/10.1007/BF00176014, 1993.
Günther, H.: NMR Spectroscopy: Basic Principles, Concepts and Applications in Chemistry, 3rd edition, Wiley, ISBN 978-3-527-33000-3, 2013.
Hierso, J.-C.: Indirect nonbonded nuclear spin–spin coupling: a guide for the recognition and understanding of “through-space” NMR J constants in small organic, organometallic, and coordination compounds, Chem. Rev., 114, 4838–4867, https://doi.org/10.1021/cr400330g, 2014.
Juszewski, K., Gronenborn, A. M., and Clore, G. M.: Improving the packing and accuracy of NMR structures with a pseudopotential for the radius of gyration, J. Am. Chem. Soc., 121, 2337–2338, https://doi.org/10.1021/ja9843730, 1999.
Kimber, B. J., Feeney, J., Roberts, G. C. K., Birdsall, B., Griffiths, D. V., Burgen, A. S. V., and Sykes, B. D.: Proximity of two tryptophan residues in dihydrofolate reductase determined by 19F NMR, Nature, 271, 184–185, https://doi.org/10.1038/271184a0, 1978.
Kruse, H., Mrazikova, K., D'Ascenzo, L., Sponer, J., and Auffinger, P.: Short but weak: the Z-DNA lone-pair…π conundrum challenges standard carbon van der Waals radii, Angew. Chem. Int. Ed., 59, 16553–16560, https://doi.org/10.1002/anie.202004201, 2020.
Lu, T., Zhang, J., Chen, J., Gou, Q., Xia, Z., and Feng, G.: Structure and non-covalent interactions of 1,3-difluoropropane and its complex with water explored by rotational spectroscopy and quantum chemical calculations, J. Chem. Phys., 150, 064305, https://doi.org/10.1063/1.5079564, 2019.
MacKenzie, K. R., Prestegard, J. H., and Engelman, D. M.: Leucine sidechain rotamers in a glycophorin A transmembrane peptide as revealed by three-bond carbon–carbon couplings and 13C chemical shifts, J. Biomol. NMR, 7, 256–260, https://doi.org/10.1007/BF00202043, 1996.
Maleckis, A., Abdelkader, E. H., Herath, I. D., and Otting, G.: Synthesis of fluorinated leucines, valines and alanines for use in protein NMR, Org. Biomol. Chem., 20, 2424–2432, https://doi.org/10.1039/D2OB00145D, 2022.
Marstokk, K.-M. and Møllendal, H.: Structural and conformational properties of 1,3-difluoropropane as studied by microwave spectroscopy and ab initio calculations, Acta Chem. Scand., 51, 1058–1065, https://doi.org/10.3891/acta.chem.scand.51-1058, 1997.
Orton, H. W., Qianzhu, H., Abdelkader, E. H., Tan, Y. J., Habel, E. I., Frkic, R. L., Jackson, C. J., Huber, T., and Otting, G.: Through-space scalar 19F–19F couplings between fluorinated non-canonical amino acids for the detection of specific contacts in proteins, J. Am. Chem. Soc., 143, 19587–19598, https://doi.org/10.1021/jacs.1c10104, 2021.
Otting, G.: NMR spectra of GB1 produced with fluorinated valine analogues along with spectra of the wild-type reference, Zenodo [data set], https://doi.org/10.5281/zenodo.17082808, 2025.
Qianzhu, H., Abdelkader, E. H., Herath, I. D., Otting, G., and Huber, T.: Site-specific incorporation of 7-fluoro-L-tryptophan into proteins by genetic encoding to monitor ligand binding by 19F NMR spectroscopy, ACS Sens., 7, 44–49, https://doi.org/10.1021/acssensors.1c02467, 2022.
Qianzhu, H., Abdelkader, E. H., Otting, G., and Huber, T.: Genetic encoding of fluoro-L-tryptophans for site-specific detection of conformational heterogeneity in proteins by NMR spectroscopy, J. Am. Chem. Soc., 146, 13641–13650, https://doi.org/10.1021/jacs.4c03743, 2024.
Qianzhu, H., Tan, Y. J., Abdelkader, E. H., Huber, T., and Otting, G.: Genetic encoding of fluorinated analogues of phenylalanine for 19F-NMR spectroscopy: detection of conformational heterogeneity in flaviviral NS2B-NS3 proteases, ACS Sens., 10, 3152–3161, https://doi.org/10.1021/acssensors.5c00432, 2025.
Tan, Y. J., Abdelkader, E. H., Tarcoveanu, E., Maleckis, A., Nitsche, C., and Otting, G.: (2S,4S)-5-Fluoroleucine, (2S,4R)-5-fluoroleucine, and 5,5'-difluoroleucine in E. coli PpiB: protein production, 19F NMR, and ligand sensing enhanced by the γ-gauche effect, Biochemistry, 63, 1376–1387, https://doi.org/10.1021/acs.biochem.4c00080, 2024.
Tan, Y. J., Abdelkader, E. H., Herath, I. D., Maleckis, A., and Otting, G.: Inter-residue through-space scalar 19F–19F couplings between CH2F groups in a protein, Magn. Reson., 6, 131–142, https://doi.org/10.5194/mr-6-131-2025, 2025.
Tonelli, A. E. and Schilling, F. C.: 13C NMR chemical shifts and the microstructure of polymers, Acc. Chem. Res., 14, 233–238, https://doi.org/10.1021/ar00068a002, 1981.
Tonelli, A. E., Schilling, F. C., and Cais, R. E.: 19F NMR chemical shifts and the microstructure of fluoro polymers, Macromolecules, 15, 849–853, https://doi.org/10.1021/ma00231a031, 1982.
Williamson, K. L., Hus, Y.-F. L., Hall, F. H., Swager, S., and Coulter, M. S.: Dihedral angle and bond angle dependence of vicinal proton–fluorine spin–spin coupling, J. Am. Chem. Soc., 90, 6717–6722, https://doi.org/10.1021/ja01026a028, 1968.
Worley, B., Richard, G., Harbison, G. S., Powers, R.: 13C NMR reveals no evidence of n–π∗ interactions in proteins, PLOS One, 7, e42075, https://doi.org//10.1371/journal.pone.0042075, 2012.
Wu, D., Tian, A., and Sun, H.: Conformational properties of 1,3-difluoropropane, J. Phys. Chem. A, 102, 9901–9905, https://doi.org/10.1021/jp982164w, 1998.