the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Apr 2024
| 19 Apr 2024
Deuteration of proteins boosted by cell lysates: high-resolution amide and Hα magic-angle-spinning (MAS) NMR without the reprotonation bottleneck
Federico Napoli
Jia-Ying Guan
Charles-Adrien Arnaud
Pavel Macek
Hugo Fraga
Cécile Breyton
Amide-proton-detected magic-angle-spinning NMR of deuterated proteins has become a main technique in NMR-based structural biology. In standard deuteration protocols that rely on D2O-based culture media, non-exchangeable amide sites remain deuterated, making these sites unobservable. Here we demonstrate that proteins produced with a H2O-based culture medium doped with deuterated cell lysate allow scientists to overcome this “reprotonation bottleneck” while retaining a high level of deuteration (ca. 80 %) and narrow linewidths. We quantified coherence lifetimes of several proteins prepared with this labeling pattern over a range of magic-angle-spinning (MAS) frequencies (40–100 kHz). We demonstrate that under commonly used conditions (50–60 kHz MAS), the amide 1H linewidths with our labeling approach are comparable to those of perdeuterated proteins and better than those of protonated samples at 100 kHz. For three proteins in the 33–50 kDa size range, many previously unobserved amides become visible. We report how to prepare the deuterated cell lysate for our approach from fractions of perdeuterated cultures which are usually discarded, and we show that such media can be used identically to commercial media. The residual protonation of Hα sites allows for well-resolved Hα-detected spectra and Hα resonance assignment, exemplified by the de novo assignment of 168 Hα sites in a 39 kDa protein. The approach based on this H2O/cell-lysate deuteration and MAS frequencies compatible with 1.3 or 1.9 mm rotors presents a strong sensitivity benefit over 0.7 mm 100 kHz MAS experiments.
- Article
(6501 KB) - Full-text XML
-
Supplement
(9541 KB) - BibTeX
- EndNote





The deuteration of proteins for NMR studies is a very important technique and has greatly expanded the scope of biomolecular NMR. In solution-state NMR, overall Brownian motion modulates the relaxation-active interactions, in particular the strong 1H–1H dipolar interactions and chemical-shift anisotropy (CSA), and when this Brownian motion is slow, which is the case for large proteins, the resulting decay of coherences is fast. This rapid relaxation and the associated line broadening represent a fundamental barrier to the size of proteins that can be studied. This barrier can be overcome by using highly deuterated proteins, where only a few sites, e.g., amides, methyls, or aromatic sites, bear a 1H spin, while all the rest are deuterated (2H). Together with transverse-relaxation optimized pulse sequences tailored for amides (Pervushin et al., 1997), methyls (Tugarinov et al., 2003), or aromatics (Pervushin et al., 1998), deuteration has alleviated the size limitations of solution-state NMR and has made proteins in the megadalton range accessible to site-resolved studies of dynamics and interactions.
In magic-angle-spinning (MAS) solid-state NMR, overall molecular tumbling that dominates relaxation in solution is absent, and the transverse relaxation is inherently slow. However, the dipole–dipole interactions are not perfectly averaged by magic-angle spinning, and these interactions lead to a rapid decay of the observable spin coherences. Traditionally, biomolecular MAS NMR has, thus, focused on observing 13C rather than 1H. The smaller gyromagnetic ratio of 13C and accordingly the smaller dipolar couplings lead to a more complete averaging of the dipolar couplings already at lower MAS frequency. However, it also comes with inherently lower detection sensitivity than 1H. In order to achieve narrow 1H linewidths, diluting the network of 1H–1H dipolar interactions by deuteration is a well-established method. Deuteration with sparse introduction of 1H nuclei only at, for example, amide (1HN), Hα, methyl, or aromatic sites, has rapidly established itself as a standard technique in biomolecular MAS NMR. The higher detection sensitivity of 1H translates to smaller sample amounts needed. Pioneering experiments, e.g., by the Reif group, used perdeuterated proteins in which only a fraction (e.g., 10 %) of the amides are protonated, thereby sacrificing signal intensity, at MAS frequencies of 20 kHz, i.e., with 3.2 mm large rotors and ca. 30 mg of material (Chevelkov et al., 2006). Faster MAS with smaller-diameter rotors allows scientists to introduce more protons while keeping narrow linewidths; 100 % amide protonation with MAS frequencies of 40–60 kHz, i.e., smaller diameter rotors of either 1.3 mm (ca. 2.5 mg material) or 1.9 mm (ca. 13 mg), has become widespread (Lewandowski et al., 2011; Zhou et al., 2007). Akin to solution-state NMR, amide-1H-detected MAS NMR experiments are employed for many aspects of protein structure and dynamics studies: they are used to obtain backbone resonance assignment (Barbet-Massin et al., 2014; Fraga et al., 2017; Xiang et al., 2015; Stanek et al., 2020; Andreas et al., 2015; Klein et al., 2022), determine structures from 1HN–1HN distances (Retel et al., 2017; Zhou et al., 2007; Knight et al., 2012; Linser et al., 2011b; Najbauer et al., 2022; Gauto et al., 2019a; Schubeis et al., 2020) or long-distance restraints to paramagnetic (Knight et al., 2012) or 19F (Shcherbakov et al., 2019) sites, or to characterize protein dynamics (Napoli et al., 2023; Singh et al., 2019; Bonaccorsi et al., 2021; Good et al., 2014; Lewandowski, 2013; Chevelkov et al., 2009) (We refer the reader to excellent recent reviews of 1H-detected MAS NMR and deuteration (Le Marchand et al., 2022; Vasa et al., 2018; Reif, 2021).) Very high MAS frequencies available today with 0.5 or 0.7 mm rotors (up to 150 kHz) alleviate the need for deuteration, and protonated proteins are sufficient for structure determination or dynamics studies (Agarwal et al., 2014; Andreas et al., 2016; Lamley et al., 2014; Bougault et al., 2019; Cala-De Paepe et al., 2017). However, the absolute detection sensitivity drops substantially due to the smaller rotor volume; for example, using a 0.7 mm rotor instead of a 1.3 mm rotor, each close to its respective maximum MAS frequency, leads to a loss of at least a factor of 2.5 in sensitivity (or 6 in experimental time) (Le Marchand et al., 2022). Thus, for many practical applications, the use of deuterated proteins with MAS frequencies in the 40–60 kHz range (1.9 or 1.3 mm rotors) is still a preferable method.
Deuterated proteins for amide-detected experiments are commonly produced by bacterial overexpression in a minimal growth medium (Gardner and Kay, 1998) (M9 medium (Anderson, 1946)) prepared with D2O. In the following, we refer to this method of deuteration in D2O-based growth media as perdeuteration. As the amide hydrogen atoms are chemically exchangeable, those accessible to the solvent are replaced with the 1H isotope when the perdeuterated protein is placed in H2O during purification and subsequent measurement. This amide reprotonation is, however, often incomplete, in particular in the core of large proteins, which leads to lack of information for these parts of the structure. As a consequence of the incomplete back-exchange of amide sites, many structurally important probes are invisible. For example, in the structure determination of TET2 (Gauto et al., 2019a) for a buried β sheet, only very few distance constraints were available.
One option to circumvent the need for reprotonation of amide sites is to focus on Hα sites instead. An elegant approach has been proposed by the Andreas group, which starts from a commercial growth medium that contains a mix of deuterated amino acids and makes use of transaminases to protonate the Hα positions (Tekwani et al., 2019), in an amino acid-type-dependent manner. Overexpression is then performed with this mix of deuterated Hα-protonated amino acids. The method, named “alpha proton exchange by transamination” (α-PET), has been shown to generate proteins that are protonated predominantly at the α sites. A potential drawback is, however, that Hα sites generally have narrower signal dispersion than amide sites, such that resolution is often lower. Moreover, dynamics studies using the Cα site are more complicated, because the 13C–13C couplings render any quantitative analysis of relaxation complex (if not impossible); dynamics studies using 15N sites are preferable.
Denaturation of the perdeuterated protein in H2O, followed by refolding, is one viable route to achieve complete reprotonation of amide sites in perdeuterated proteins (Gardner et al., 1998). However, it is often difficult or even impossible to find suitable conditions in which the protein retrieves its native structure.
An alternative way to obtain (at least partly) deuterated proteins in which all exchangeable hydrogen sites bear a 1H isotope is to perform the deuteration in H2O while providing the building blocks that serve for protein synthesis in a deuterated form. A simple approach is to use deuterated D-glucose as sole carbon source in H2O-based minimal (M9) medium; this approach has been proposed for MAS NMR and termed inverse fractional deuteration (iFD) (Medeiros-Silva et al., 2016). As the entire biosynthesis of amino acids from glucose takes place in H2O, this approach results in relatively low deuteration levels; in particular, the α sites have been reported to be protonated to 88 %–100 % for all reported amino acids (Table S2 of Medeiros-Silva et al., 2016). Due to the low overall deuteration, the coherence lifetimes of the amide 1H spins in the iFD scheme are close to those of protonated samples; that is, the gain in resolution with this sparse deuteration is limited (Cala-De Paepe et al., 2017).
Asami et al. have proposed another approach, which primarily aims to introduce aliphatic protons in a sparse manner but which also results in partial amide protonation. The approach, termed “random adjoining protonation”, consists of growing bacteria in a mixture of H2O and D2O and protonated 13C glucose (Asami et al., 2010, 2012). The aim is not to achieve the highest possible deuteration at non-amide sites but rather some level of aliphatic protonation for detection of, for example, methyl sites. Thus, the approach is not optimized for amide detection.
Using deuterated amino acids in the growth medium promises to result in more complete deuteration. Löhr et al. grew cultures in H2O-based medium with a commercial deuterated algal lysate (without adding glucose) for solution-state NMR study of a 35 kDa large protein (Löhr et al., 2003). A similar strategy has been reported by the Wand group (O'Brien et al., 2018). This approach has not been reported for MAS NMR, to our knowledge. A significant drawback of this method is the price. The lysate that Löhr et al. used has a current list price of USD 6500 for 1 L of culture (December 2023), which may be considered prohibitively expensive in many cases. Moreover, the amino acids are partly reprotonated due to the action of transaminases, and this protonation largely varies depending on the amino acid type; the residual protonation at α sites can reach 80 % (Löhr et al., 2003). Besides bacterial expression, cell-free protein production in H2O with deuterated amino acids can be performed. When used with transaminase inhibitors, the level of α protonation can be maintained below ca. 10 % (Imbert et al., 2021; Xuncheng et al., 2011). However, few laboratories have established cell-free production, and this approach is less readily accessible than bacterial overexpression.
Here, we propose an approach for producing highly (≥80 %) deuterated proteins in H2O-based M9 medium with deuterated D-glucose supplemented with deuterated cell lysate. We find that 2 g of cell lysate powder added to the medium is sufficient to reach 80 % overall deuteration level, and this level is ca. 4 times higher than the one obtained with the previously proposed iFD approach (Medeiros-Silva et al., 2016). While commercial deuterated media can be used, we report the straightforward preparation of a home-made cell lysate from what is usually discarded from perdeuterated bacterial protein production, and we show that its properties are indistinguishable from a commercial medium.
Moreover, we investigate the 1H coherence lifetimes of several proteins produced with this H2O/M9/cell-lysate deuteration scheme, and we compare them to those of perdeuterated and fully protonated protein samples over a range of MAS frequencies up to 100 kHz. Most importantly, we report that the amide 1H linewidths of the H2O/M9/cell-lysate-deuterated samples are not significantly different from those of perdeuterated samples (although the coherence lifetimes, i.e., the homogeneous linewidths (Le Marchand et al., 2022), of the perdeuterated samples are favorable). Using three proteins with molecular weights ranging from 33 to 50 kDa in 1.3 or 1.9 mm rotors (38–55 kHz MAS frequency), we show that our approach retrieves many amide signals that were lost in a standard perdeuteration approach. For the 12×39 kDa large protein assembly TET2, we obtained 40 % more amide assignments than with the previous perdeuteration approach, most of which was in the hydrophobic core of the protein. We show furthermore that the residual unwanted protonation of α sites can be exploited in 3D and 4D experiments that edit the Hα frequency. We obtained 168 Hα chemical-shift assignments, which is close to the number obtained from a fully protonated sample at 100 kHz MAS.
2.1 Deuteration of proteins in H2O-based cultures with deuterated algal lysate
The deuteration level and deuteration pattern (i.e., which sites are deuterated) are essential for the resolution in amide-hydrogen-based NMR MAS spectra. We first quantified the overall deuteration level that can be reached using H2O-based bacterial cultures producing the 33.55 kDa protein MalDH, by determining the intact protein mass with mass spectrometry (Fig. 1a). When only deuterated glucose is used (at 2 g L−1 of culture; this corresponds to the previously proposed “iFD” approach (Medeiros-Silva et al., 2016)), the resulting protein is deuterated to ca. 25 % overall. The addition of the deuterated algal lysate-derived complex labeling medium increases the deuteration level in a concentration-dependent manner: upon addition of ca. 2 g L−1 of ISOGRO® powder to the medium, a plateau level of ca. 80 % deuteration is reached. Doubling the amount of ISOGRO® added to the medium to 4 g L−1 has only a very small effect, such that we consider 2 g L−1 as a sufficient amount. For brevity, we refer in the remainder of this text to the deuteration that uses H2O culture medium with deuterated M9 components (including 2 g L−1 deuterated D-glucose) and deuterated cell lysate (at 2 g L−1) as the “H2O/M9/lysate” sample. With lysate, we refer either to the commercial complex growth medium (such as ISOGRO®) or an in-house-produced variant of it (see below). We refer to “perdeuteration” as the approach using D2O-based M9 medium using 2 g L−1 deuterated D-glucose. In all cases, the final NMR samples are in a buffer composed of H2O.
Having quantified the overall deuteration level, we investigated more closely the pattern of the residual protonation. To this end, we have produced the 8.6 kDa protein ubiquitin with different labeling patterns: either no deuteration (u-[13C,15N]), perdeuteration (u-[2H,13C,15N]), or deuteration with the H2O/M9/lysate approach proposed herein. For the latter, the M9 culture medium was in H2O and included 2 g L−1 2H,13C glucose; 1 g L−1 15N ammonium; and 2 g L−1 2H,13C,15N ISOGRO® powder. We measured the deuteration level with 1H–13C heteronuclear single-quantum coherence (HSQC) spectra, which provides simultaneously the deuteration levels for all aliphatic sites.
Figure 1b and c show the protonation levels in aliphatic side chains and at Hα positions. Marked differences were found for Hα deuteration of different amino acid types, qualitatively similarly to previously reported data (Löhr et al., 2003). For example, while Glu and Phe have a high incorporation of 1Hα, Lys and Ala have a higher deuteration level. Positions further out in the side chain have a higher deuteration level. Overall, our data show that there is significant variation in the deuteration level, which we ascribe to the activity of transaminases that differs for the types of amino acids. While the residual protonation may also be of possible use (namely, for 1H detection of aliphatic hydrogen sites), as we will show below the presence of these 1H spins in the vicinity to amide protons will likely accelerate the coherence decay of amide 1H spins. This question of coherence lifetimes and linewidths is addressed in a later section in this article.
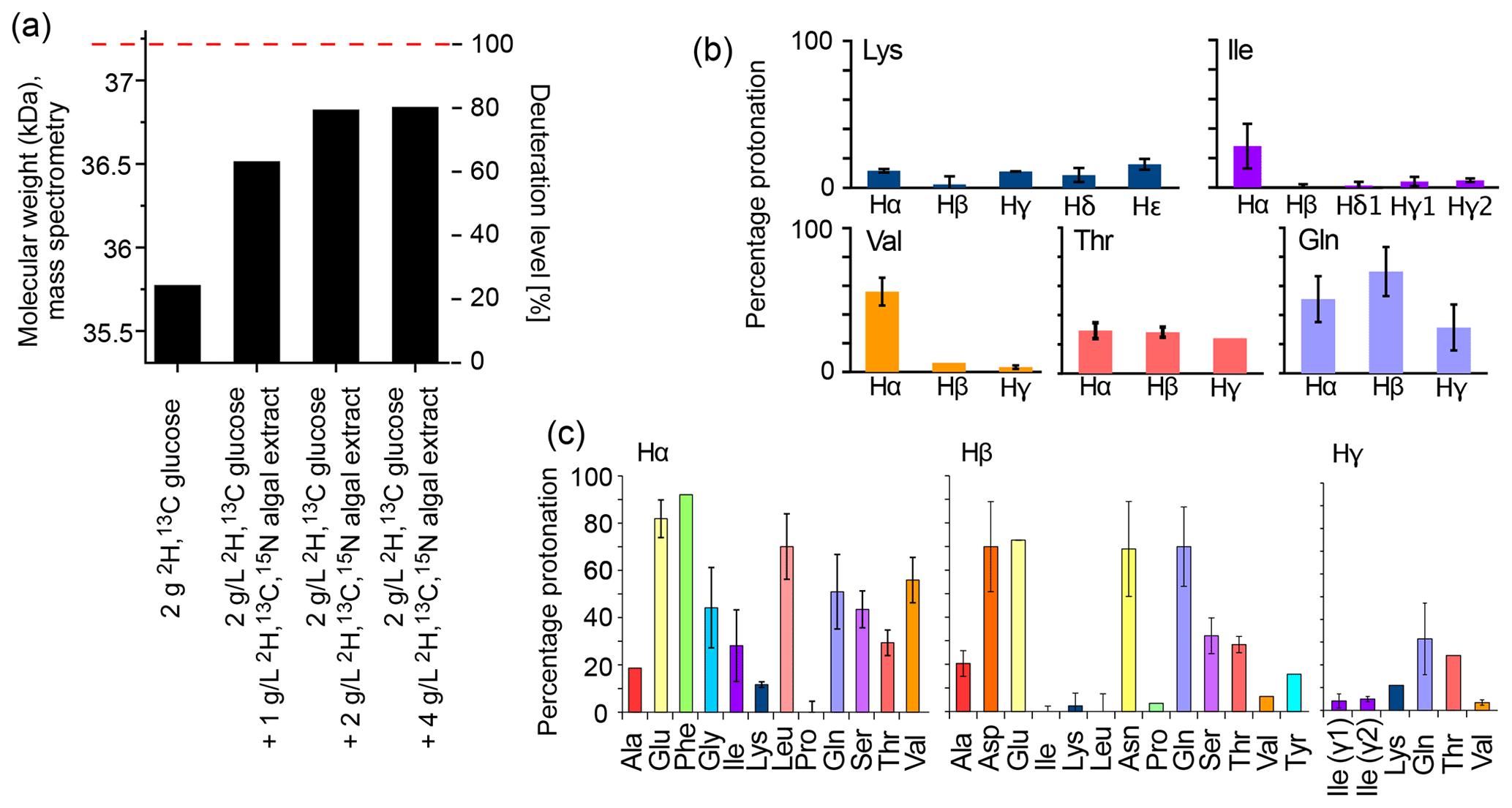
Figure 1Deuteration using H2O-based medium doped with algal lysate. (a) Deuteration level of Ignicoccus islandicus MalDH (33.55 kDa) expressed in H2O with either 2H,13C glucose only (2 g per liter of culture) and 15NH4 or with additional use of 2H,13C,15N algal extract (ISOGRO®) at three different concentrations (1, 2, 4 g per liter of culture). The reported molecular-weight values are from intact mass spectrometry. The dashed red line indicates the theoretical molecular weight of fully deuterated MalDH (assuming all exchangeable hydrogens are 1H). (b, c) Residual protonation level for aliphatic sites, determined by solution-state NMR of a sample of ubiquitin produced in H2O-based M9 medium supplemented with 2H,13C,15N algal extract (2 g L−1). Amino acid types missing in this plot are either not present in ubiquitin, excluding the possibility for quantification (Trp), or are not visible or unresolved (Met, His, Arg).
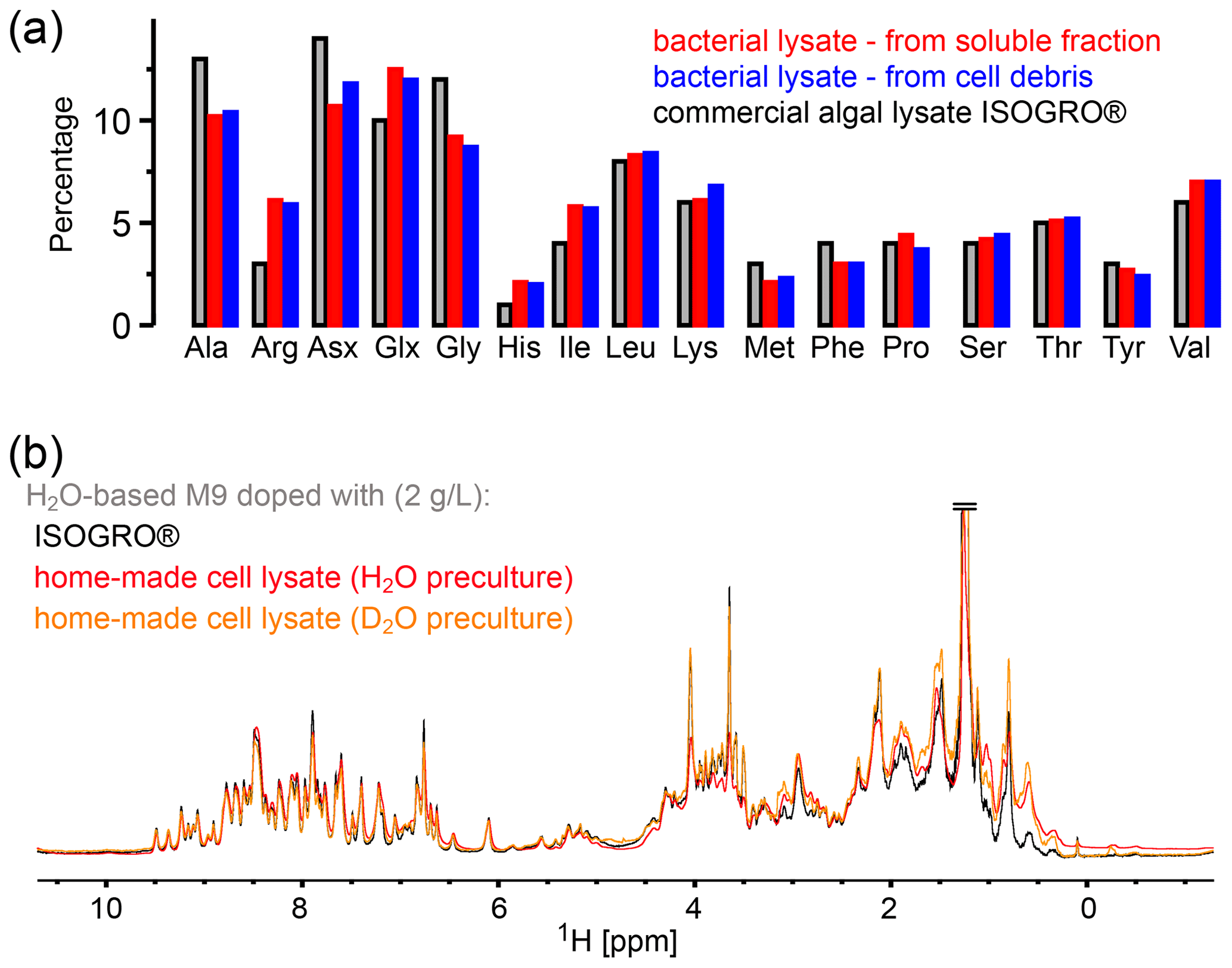
Figure 2Comparison of the amino acid composition in a home-made bacterial extract with the one in ISOGRO®. (a) The amino acid composition of ISOGRO®, as provided by the manufacturer, is shown as grey bars; the values represent the percentage (weight) of each amino acid in the powder. Data of the home-made preparation from bacterial perdeuterated cultures are shown in blue and red. The preparation made from the soluble-protein fraction, i.e., contaminant proteins from a protein purification, is shown in red. The preparation made from the insoluble fraction after cell lysis is shown in blue. Asn and Gln are indistinguishable from Asp and Glu with this method. Cys and Trp are destroyed during the processing steps of the analysis. (b) 1D 1H spectra of ubiquitin deuterated in a H2O-based medium supplemented with either commercial algal lysate (black) or the home-made lysate, prepared from the contaminant proteins. The precultures have been made either in H2O or D2O, as indicated. The similarity of the spectra confirms that commercial algal lysates and bacterial lysates produce similar labeling patterns, as expected from the very similar distribution of amino acids shown in panel (a).
2.2 Straightforward preparation of an isotope-labeled cell lysate as an alternative to commercial media
Having shown that a commercial deuterated algal extract (e.g., ISOGRO® from Sigma-Aldrich, Celtone® from Cambridge Isotope Laboratories, or SILEX® media from Silantes) allows for obtaining high deuteration levels in H2O-based bacterial expression, we have investigated the possibility to prepare such a medium in house. In many laboratories, deuterated proteins are prepared from Escherichia coli cultures grown in D2O-based M9 media. While the D2O from such cultures is often reused, after distillation, the contaminant proteins and the insoluble pellet are usually discarded. We have set up a very simple method to make use of this deuterated material, and we use it in the same way as the commercial lysate to deuterate proteins.
In principle, different parts may be recycled after the protein of interest is purified: either the soluble contaminant proteins or the insoluble debris. We have explored both possibilities. The soluble contaminant proteins were retained during the Ni-affinity purification step; the insoluble fraction was simply the part that was in the pellet after a centrifugation following cell lysis. In both cases, the sample was treated with phosphoric acid to hydrolyze the peptide bonds. The pH was neutralized by NaOH, and the sample was cleared by ultracentrifugation and then lyophilized. We have obtained ca. 375 mg of lyophilized powder from 1 L of culture using the soluble fraction and ca. 900 and 1400 mg (two independent samples) from the insoluble debris. We have analyzed the amino acid composition of these powders by amino acid analysis. Figure 2 compares the relative amino acid composition of these two samples to the composition of the commercial ISOGRO® medium. The composition is similar, which suggests that the cell lysate from bacteria can be used in the same manner as the commercial algal lysate.
To test this assumption, we have produced samples of ubiquitin with either deuterated commercial algal lysate (ISOGRO®) or the home-made bacterial lysate described above (2 g L−1 each), and we recorded NMR spectra (Fig. 2b). The spectra show a very similar level of deuteration, supporting the argument that bacterial cell lysate is a viable alternative to commercial ones. Small differences are observed, e.g., in the methyl region. However, these differences do not seem to be fundamental: in fact, when comparing different productions (even from the same batch of lysate), the intensities of aliphatic sites in 1D spectra vary in a similar manner as the variations seen in Fig. 2b. Given that the deuteration levels of, for example, methyls are of only a few percent (see Fig. 1b), even a factor of 2 in the deuteration level of a given methyl will hardly change the absolute deuteration level. The details will depend on the deuteration level of the culture from which the cell lysate has been made. The bacterial cell lysate comes essentially free of additional cost when perdeuterated proteins are made, considering that it uses fractions that are usually discarded. However, with a yield of ca. 1 g L−1 of culture, ca. 2 L of a deuterated culture is needed to prepare 1 L of H2O/M9/cell-lysate-deuterated sample with good deuteration (see Fig. 1a). Overall, we have shown that bacterial lysate and commercial algal lysate can be used interchangeably; this assessment is also confirmed by an application of the home-made bacterial lysate to the MAS NMR study of the pb6 protein assembly (see below).
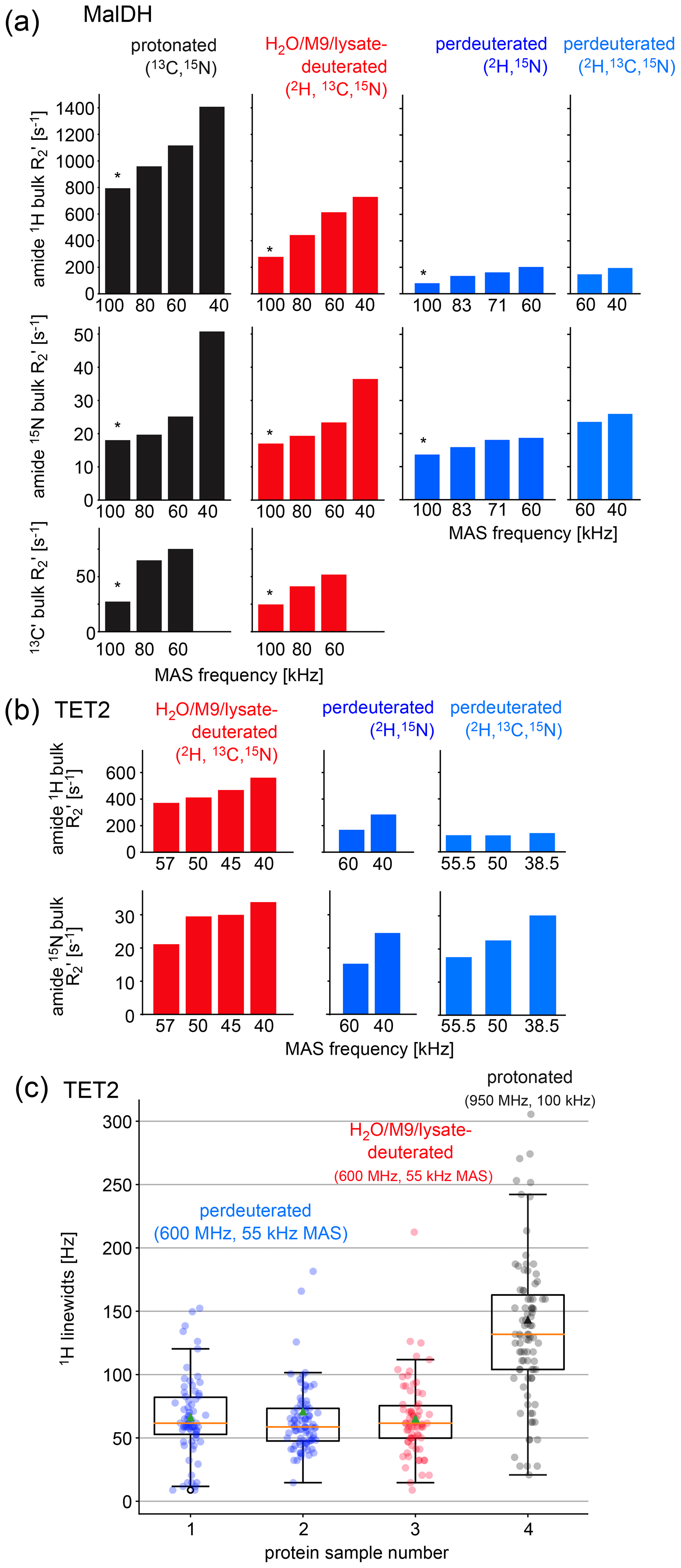
Figure 3Investigation of coherence lifetimes and linewidths in differently labeled samples of two proteins. (a) Fitted R2′ decay rate constants for four differently labeled samples of MalDH as indicated. The decay curves for representative experiments, indicated with an asterisk, are shown in Fig. S1. All data were recorded at a B0 field strength corresponding to 700 MHz 1H Larmor frequency in a 0.7 mm probe. (b) Similar data for the TET2 protein. (c) 1H linewidths of TET2 in deuterated samples produced in D2O-based M9 medium (blue), produced in a H2O-based M9 medium doped with 2 g L−1 deuterated algal extract (red) or fully protonated (black). Spectra are shown in Fig. S2.
2.3 Coherence lifetimes in H2O/M9/cell-lysate-deuterated samples
Two key parameters we are interested in are the coherence lifetimes (T2′) and the linewidths. The linewidth contains contributions from the coherence lifetime (the so-called homogeneous contribution), but additional sample inhomogeneity also contributes (Le Marchand et al., 2022). Which of these contributions dominates depends on the sample and the conditions (in particular the MAS frequency). To compare the H2O/M9/cell-lysate samples with perdeuterated and protonated samples, we have, thus, analyzed both the homogeneous contribution and the apparent linewidths in two large test proteins, TET2 (39 kDa monomer size, assembling to a dodecamer) and MalDH (33.55 kDa monomer size, assembling to a tetramer). To quantify the homogeneous contribution, we have measured the coherence decay using spin-echo experiments. The experiments applied a spin echo (τ – 180° pulse – τ) element to either 1H, 15N, or 13CO coherence, embedded into a heteronuclear correlation experiment with 1H detection. We compared samples prepared with full deuteration (produced in D2O medium), with samples deuterated in H2O medium doped with deuterated algal lysate, and finally a fully protonated sample. As shown above, these three samples correspond to different levels of deuteration. In all cases, the proteins were in H2O buffer; that is, all the exchangeable sites are protonated to either 100 % (for the two types of samples grown in H2O medium) or to a level that depends on the accessibility of the given amide site. Because the importance of deuteration depends on the MAS frequency (Le Marchand et al., 2022), we have performed the measurements at various MAS frequencies up to 100 kHz (Fig. 3).
As expected from the deuteration level, the 1H coherence lifetimes of the samples deuterated with algal lysate in H2O (red in Fig. 3) are in between those of perdeuterated samples (blue) and the fully protonated sample (black). Of note is that, even at the highest MAS frequency used in this study, 100 kHz, the 1H coherence lifetime of the fully protonated sample is more than 3 times shorter than the one of the sample deuterated with algal medium. Moreover, the 1H coherence lifetime of the fully protonated sample spinning at 100 kHz is significantly (more than 20 %) shorter than the one of the algal-medium-deuterated sample spinning at 60 kHz. This is an important realization, because in order to be able to spin at 100 kHz, one needs to use smaller rotors and sacrifice about a factor of 5 in sample amount, which translates to a severe sensitivity penalty of 100 kHz MAS experiments. As expected from the deuteration level study above, the perdeuterated sample (placed in H2O) has longer 1H coherence lifetimes (lower R2′). Moreover, in absolute terms, the MAS dependency is less pronounced for the deuterated sample than the protonated one.
The decay of heteronuclear (amide 15N, carbonyl 13C′) coherences depends much less on the deuteration level (Fig. 3). It depends significantly on the 1H decoupling scheme and decoupling power used, and the optimum decoupling scheme and decoupling power generally depends on the MAS frequency. To generate a consistent data set, we have used low-power 1H WALTZ-16 (Shaka et al., 1983) decoupling (ca. 10 kHz), which is not the best-performing scheme at 40 kHz (data not shown). Therefore, these data are to be taken with a grain of salt, and the main message from them is that 15N and 13C′ decay is much less dependent on the labeling pattern than 1H coherence lifetimes. Of note is that the experiments performed here did not use 2H decoupling; the rationale is that most probes are not equipped with a 2H coil.
2.4 Linewidths in H2O/M9/cell-lysate-deuterated samples at 55 kHz MAS are similar to perdeuteration and better than protonated samples at 100 kHz
The linewidths observed in spectra contain additional inhomogeneous contributions due to sample heterogeneity, inhomogeneity of the magnetic field, and anisotropic bulk magnetic susceptibility (Le Marchand et al., 2022; Linser et al., 2014). The R2′ rate constants discussed above do not comprise these effects. We have measured the linewidths in a series of 2D hNH spectra obtained with TET2 produced either with the H2O/M9/lysate approach or by perdeuteration (Figs. 3c and S2). We find that the amide 1H linewidths in samples deuterated in H2O with deuterated algal lysate are comparable to those of proteins produced in D2O. Importantly, the linewidths of deuterated proteins at 55 kHz MAS frequency are significantly smaller than linewidths of the same protein in protonated form, spinning at 100 kHz MAS frequency.
Taken together, the above analyses showed that proteins deuterated in H2O with deuterated cell lysate (and 100 % back-exchanged in the final sample) have slightly worse spectroscopic properties (shorter 1H coherence lifetimes) than perdeuterated proteins under comparable conditions; the 1H linewidths, however, are not significantly wider.
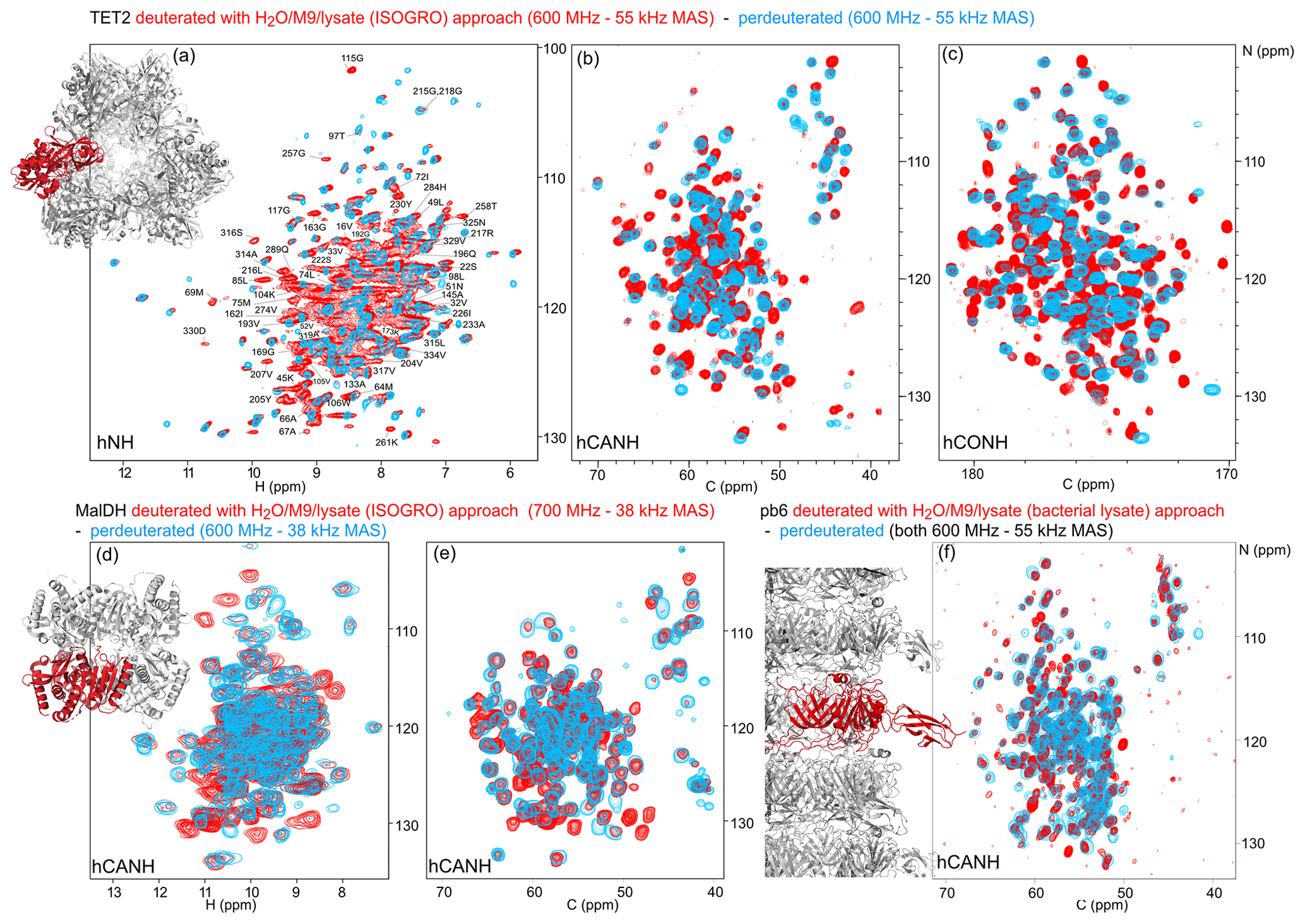
Figure 4Deuteration in H2O enhanced by amino acid mixtures from cell lysates allows for the detection of non-exchangeable amide hydrogens. Overlay of (a) hNH, (b) hCANH, (c) hCONH TET2 spectra, (d) H–N, and (e) CA–N projections of the hCANH MalDH spectra, as well as (f) hCANH pb6 spectra. (For the latter, the in-house bacterial cell lysate was used.) Spectra of samples deuterated by the H2O/M9/lysate approach are shown in red, while spectra from perdeuterated samples are shown in blue. All samples were 13C and 15N labeled. Peaks that appear to only be present in the perdeuterated sample spectra can be explained by (i) different apparent frequencies of aliased peaks (since the spectral widths and carriers are not identical in the overlaid spectra), (ii) chemical shift perturbations due to small sample-temperature differences, and (iii) different signal-to-noise ratios of individual peaks.
2.5 Application to three large proteins: retrieving the signals of non-exchangeable amide sites
To show the potential of this deuteration of proteins with full amide protonation, we have recorded three-dimensional correlation spectra of three large proteins: TET2, MalDH, and the 50 kDa large bacteriophage T5 tail protein pb6. Figure 4 shows the overlays of the hNH, hCONH, and hCANH spectra of TET2, as well as hCANH spectra of MalDH and pb6. It is apparent in all three cases that numerous peaks which are undetected in the standard perdeuteration protocol (blue) are visible in the H2O/M9/lysate-deuterated protein (red).
To gain more insight, we have analyzed in detail which amide signals are visible in TET2 with the two different deuteration approaches. TET2 is an ideal real-world application. With a 39 kDa monomer size, it is one of the largest proteins for which extensive assignments have been reported (Gauto et al., 2019a). The assignment of TET2 has been achieved with a combination of 3D and 4D 13C-detected experiments on protonated samples and 1H-detected 3D correlation experiments on perdeuterated TET2. With this, 85 % of the backbone and 70 % of the side-chain heavy atoms have been assigned in this way; 140 amide hydrogen frequencies have been assigned using the standard perdeuterated samples (see Table S1 in the Supplement). TET2 is a very stable protein, originating from a hyperthermophilic archaeon, which suggests that the protein rarely populates partially unfolded conformations that would be required to reprotonate the amides. Unfolding/refolding comes with large sample losses, such that the H2O/M9/lysate approach is ideal.
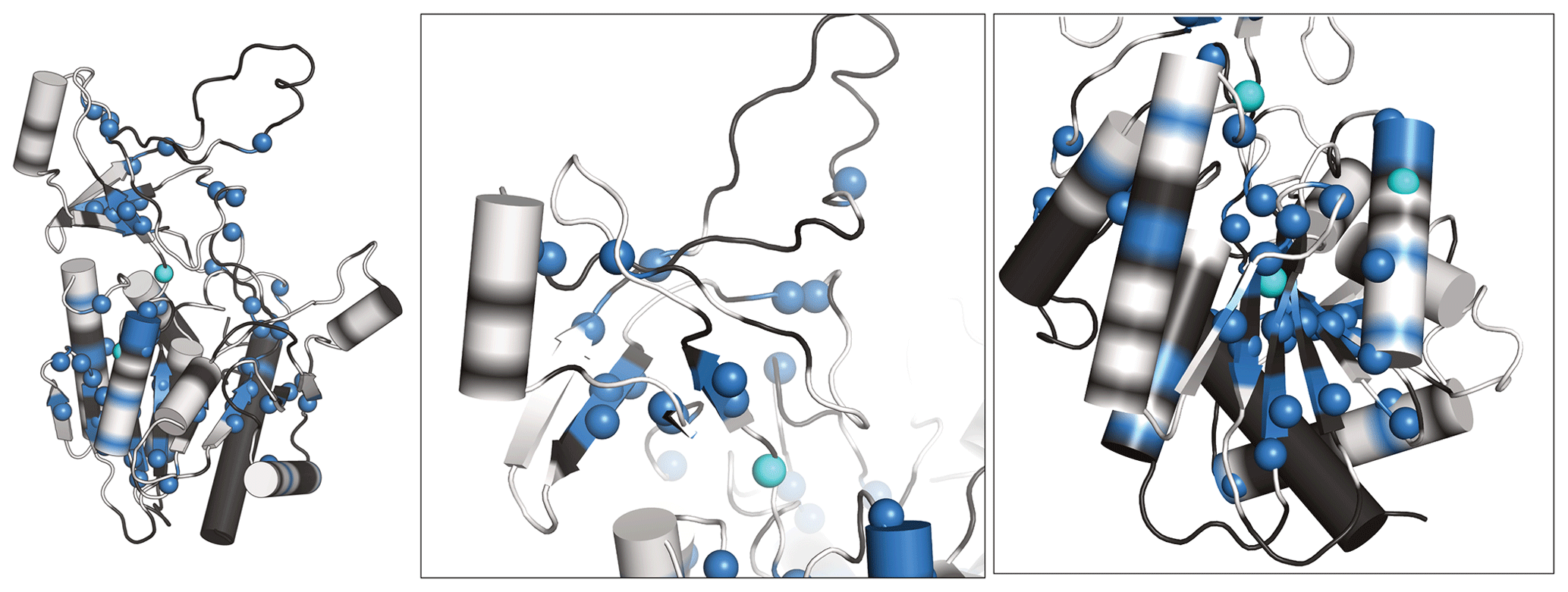
Figure 5Newly assigned amide hydrogens in TET2 deuterated in H2O shown on its monomeric structure. Amide 1H frequencies that were previously inaccessible with perdeuterated samples and that were assigned with the H2O/M9/lysate deuteration are represented as spheres on the protein structure. Blue spheres correspond to atoms that could be assigned from the spectra of the H2O/M9/lysate-deuterated sample (600 MHz 1H Larmor frequency, 55 kHz MAS), while cyan spheres are for atoms that only showed signal in the spectra of the fully protonated sample (950 MHz 1H Larmor frequency, 100 kHz MAS). Parts for which the backbone is shown in white regions correspond to those for which heavy-atom assignments have previously been reported (Gauto et al., 2019a), while black regions indicate parts with missing heavy-atom assignment. In those parts we did not attempt to get new amide assignments, as the de novo assignment of the backbone would likely require more 3D data sets. Taken together, these data highlight the possibility of detecting amide hydrogens of water-inaccessible regions in the protein. Note that the protein forms a dodecamer (Fig. 4a), and for better visibility only the monomeric subunit is shown.
We have recorded 3D assignment experiments (hCANH, hCONH) with a sample deuterated in H2O with cell lysate. These experiments allowed us to assign an additional 58 amide resonances (see Table S1). Figure 5 plots the location of these newly assigned amide resonances in the structure and shows that the majority of them are located in the β sheets of TET2 as well as in several α helices. Interestingly, also a few resonances in more exposed regions had not been assigned before and have now become assigned.
With a total of ca. 200 assigned amide hydrogens for a protein of 338 non-proline residues, there are still missing assignments left. This is in part explained by the fact that ca. 15 % of the backbone heavy-atom resonances have not been assigned by 13C-detected experiments, which is ascribed at least for some parts to extensively fast transverse relaxation due to microsecond dynamics (Gauto et al., 2022). Moreover, the somewhat limited set of 3D experiments recorded here was not sufficient to unambiguously assign all detected spin systems, as the intention of this study was to investigate the labeling scheme rather than to do an extensive resonance-assignment effort.
We investigated whether an alternative approach, namely, the use of fully protonated samples spinning at 100 kHz, would allow for the assignment of many additional resonances: hCANH and hCONH experiments allowed us to assign only three additional amide frequencies, which brings the number of assigned amide 1H sites to 201 (Table S1). We note that the sensitivity of these experiments, recorded in 0.7 mm rotors, is much lower than the one of experiments in 1.3 mm rotors, such that the use of samples deuterated in H2O represents a clear sensitivity advantage. Figure S3 shows a comparison of 2D spectra of protonated and deuterated samples.
Overall, these data exemplify how the use of proteins deuterated in H2O provides access to amide sites that are difficult to exchange, e.g., buried in the hydrophobic core.
2.6 Residual Hα protonation enables Hα-based experiments and Hα assignment
Our investigation of the deuteration pattern (Fig. 1) and, similarly, previously reported results (Löhr et al., 2003) show that there is a significant fraction of the α sites which bear a 1H spin. While this residual protonation reduces the coherence lifetimes of amide 1H spins, it can also be turned to an advantage, as the 1Hα frequency becomes available as an additional reporter.
We have exploited the residual 1Hα protonation in the H2O/M9/lysate-deuterated sample to assign 1Hα frequencies in TET2. To this end, we have recorded a 4D Hα–Cα–N–H correlation experiment with three cross-polarization (CP) steps (HACANH), at 55 kHz MAS frequency. As the Cα, N, and H frequencies are known for most residues, the assignment of the Hα frequency is straightforward with this experiment. Figure 6a shows examples from this 4D experiment. This experiment allowed for the assignment of 157 Hα resonances (Table S1).
In addition, we have recorded a complementary assignment experiment with a 13C, 15N-labeled (protonated) sample at 100 kHz MAS frequency, which correlates the Hα to the directly bonded 13Cα and the adjacent 15N (hNCAHA; Fig. 6b, grey). These two experiments both report on the Hα resonances. The 3D hNCAHA experiment, also based on three cross-polarization steps, relies on the ability to resolve 13Cα–15N correlations in two dimensions, in order to unambiguously connect the Hα frequency, while the 4D HACANH spreads the signal across three previously assigned dimensions and, therefore, is better at providing unambiguous assignments. Eleven additional Hα assignments were obtained from the experiment on the protonated samples. The assigned Hα–Cα resonances are indicated on the 2D hCH spectrum in Fig. 6c, and the location of the Hα assignments along the sequence is shown in Fig. 6d and e. Hα assignments provide additional secondary-structure information and may help to better define the secondary-structure assignments using programs such as TALOS-N (Shen and Bax, 2013); the comparison of TALOS-N results with and without the Hα assignments (Fig. S4) shows only small differences.
Taken together, while the partial protonation of α sites is in principle an unwanted by-product of the deuteration in H2O, we have shown here that the Hα nuclei can be used as an additional nucleus with good resolution, already at MAS frequency of 50–60 kHz, where fully protonated samples show poorly resolved aliphatic 1H peaks.
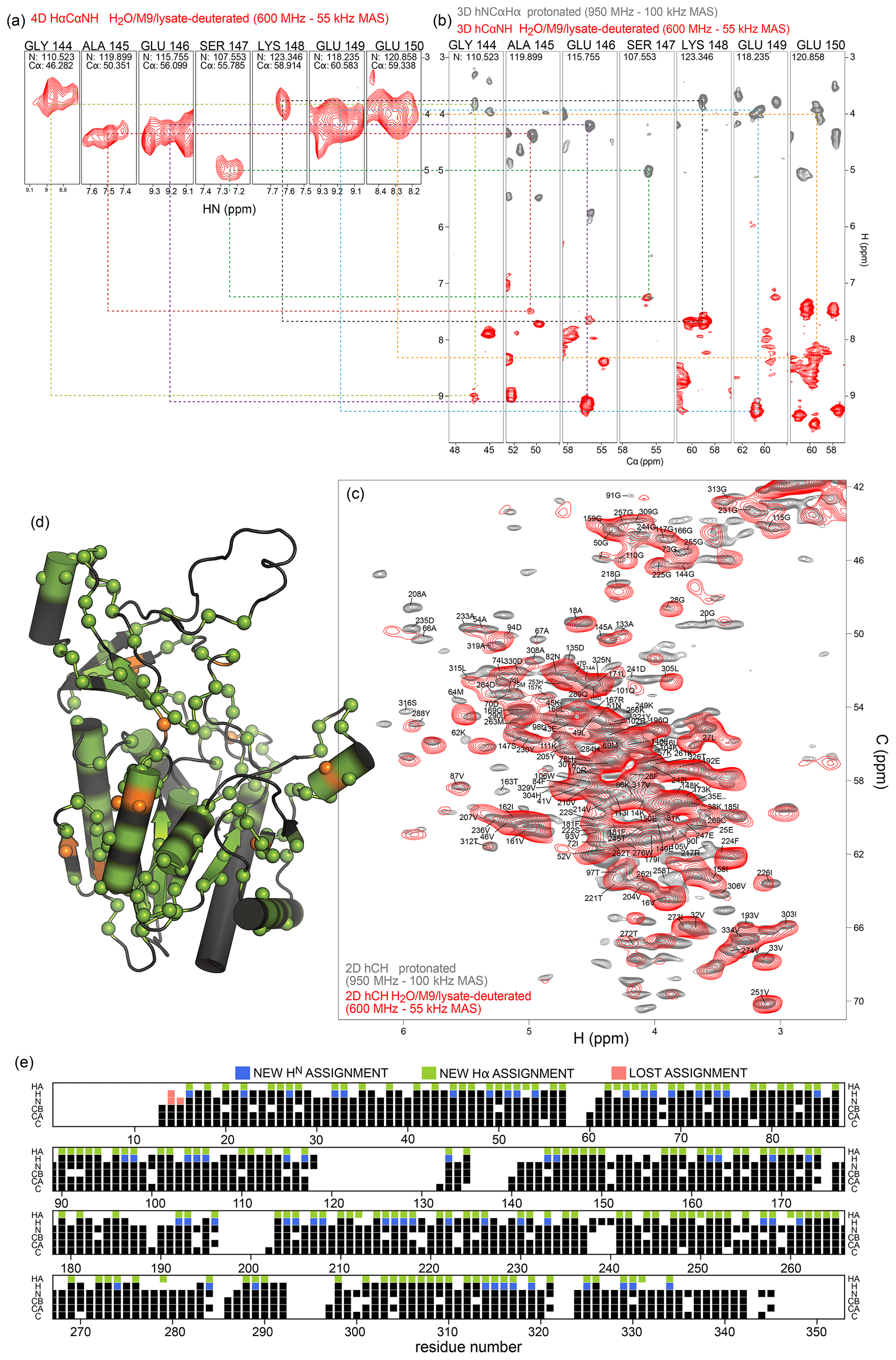
Figure 6Protein deuteration in H2O allows assignment of Hα hydrogens. (a) Example strips from 4D HACANH and (b) 3D hNCAHA and hCANH correlation experiments exploited for the assignment of Hα hydrogens. (c) Overlay of the Hα region of the hCH spectra for the fully protonated (grey) and deuterated in H2O (red) samples. The same color scheme applies to panels (a) and (b). (d) Newly assigned Hα hydrogens are represented as spheres on the protein structure. Green spheres correspond to atoms that could be assigned from the spectra of the sample deuterated in H2O, while orange spheres are for atoms that only showed signal in the fully protonated sample's spectra. Black regions indicate missing Hα assignment. (e) Sequence-based representation of TET2 resonance assignments. Newly assigned amide and Hα hydrogens are shown in blue and green, respectively. Lost assignments correspond to sites for which the automated-assignment software FLYA (Schmidt and Güntert, 2012) did not converge after adding the peak list from the H2O/M9/lysate-deuterated sample to the ones used previously for automatic assignment (Gauto et al., 2019a). Protein deuteration in H2O allows assignment of Hα hydrogens.
For the deuteration of proteins by bacterial overexpression, several approaches have been proposed which differ in the nature of the growth-media components added, as well as the pattern and amount of labeling obtained in the final protein. As a rough guide of what labeling can be expected, it is useful to keep in mind that protons from glucose tend to end up in side-chain positions, α protons tend to come from the solvent (H2O vs. D2O) due to transaminases activity, and amides are protonated to a level that corresponds to the H2O content. If bacteria do not need to synthesize amino acids from glucose because these are provided in deuterated form, then the side chains tend to be deuterated to a level close to the one of the added amino acids. With these rough guidelines in mind, one can understand that the “iFD” approach (Medeiros-Silva et al., 2016), which uses 100 % H2O and deuterated glucose, has a lower deuteration level than the H2O/lysate approach used here. The “random adjoining protonation” approach (Asami et al., 2010, 2012), which uses protonated glucose and ca. 75 % to 95 % D2O, yields a higher protonation level at side-chain positions, in particular methyls. The approach we advocate here which combines H2O, deuterated glucose, and deuterated amino acids from a lysate yields the highest deuteration level among these approaches (overall ca. 80 % deuteration), and 100 % amide protonation. Compared to the iFD method (Medeiros-Silva et al., 2016), our approach enhances the deuteration ca. 4-fold.
The deuteration level is lower for α sites and varies depending on the amino acid type, presumably due to the efficiencies of the transaminases acting on different amino acid types. Despite the substantial residual Hα protonation, which leads to somewhat shorter amide-1H coherence lifetimes (Fig. 3), the linewidths, which include inhomogeneous contributions, are similar to those of perdeuterated samples. We have demonstrated the utility of the approach for high-resolution 1H-detected MAS NMR using two large soluble protein complexes and a tube-forming protein, all of which have subunit sizes in the 33–50 kDa range. The approach will likely also become useful for membrane proteins, which have solvent-occluded parts that are often difficult to reprotonate at the amide sites (Medeiros-Silva et al., 2016).
The residual protonation of aliphatics, and in particular of α sites, can be used to obtain assignments of Hα frequencies. One may also use these residual protons for proton–proton distance measurements. A possible advantage over fully protonated samples is that the aliphatic protons are more dilute, thereby reducing dipolar truncation effects that represent a challenge for such measurements (Jain et al., 2017). A drawback of the sparseness of the aliphatic protons, however, is that the probability of observing a contact depends on the product of the populations of the two involved protons. In this respect, a specific labeling to near 100 %, such as methyl labeling, is certainly more sensitive (Huber et al., 2011; Linser et al., 2011a).
Given that the protonation level at the Hα sites varies for different amino acid types (Fig. 1), one may use Hα-detected or Hα-filtered experiments for amino acid type editing. We have not explored this possibility at this stage. A possible difficulty that needs to be addressed in this direction is the fact that, even with a uniform protonation level, peak intensities often differ vastly because of, for example, differences in transfer efficiency.
Our approach is very similar to (and conceptually the same as) the previously proposed method of deuteration in H2O which uses a bacterial growth medium (such as SILEX® or ISOGRO®) without the other M9 medium components, as proposed for solution-state NMR (Löhr et al., 2003) or for MAS NMR (Aucharova et al., 2024), published simultaneously with this article by the Linser group. There are slight variations in the amount of components used in these reports and thus the prices of the samples. It is interesting to obtain an estimate of the sample costs by considering competitive prices we obtained from different providers at the end of 2023 (D2O ca. EUR 400 per liter, 2H,13C D-glucose ca. EUR 300–400 per gram, 13C D-glucose ca. EUR 300 per gram, algal lysate 2H,13C,15N ca. EUR 200–350 per gram). These numbers result in a cost of ca. EUR 1500 per liter of culture for a sample deuterated in H2O with cell lysate as proposed here and ca. EUR 1200 per liter for a perdeuterated sample. Using more of the complex growth media (SILEX® or ISOGRO®), as proposed elsewhere, leads to somewhat higher prices (Löhr et al., 2003; Aucharova et al., 2024). The cost of the samples can further be reduced, compared to what has been used in our study: O'Brien et al. (2018) have obtained deuteration levels approaching 80 % using 2 g of algal cell extract and 1 g of unlabeled glucose per liter of culture (as opposed to 2 g L−1 deuterated glucose used here). They assumed that this quantity of glucose may be required to maintain some cellular functions of E. coli (O'Brien et al., 2018). Accordingly, the cost of the algal-lysate/H2O approach would be further reduced to EUR 400–700 per liter, which is on the order of 2-fold cheaper than a perdeuterated sample. Of course, these numbers are to be taken with caution as they vary. An additional advantage of the use of cell lysates, compared to perdeuteration, is that they often also boost protein production significantly, effectively lowering the cost of this approach further, compared to perdeuteration. The details depend very much on the protein and expression conditions.
Compared to the use of protonated samples used at very high MAS frequencies, the deuteration approach proposed here has significant advantages in terms of NMR parameters. Amide-1H linewidths obtained with protonated samples spun at 100 kHz (0.7 mm rotor) are significantly larger than those of H2O/M9/lysate-deuterated samples spun at 55–60 kHz (1.3 mm rotor) and similar to those obtained when spun at 40 kHz (1.9 mm rotor; see Fig. 3c). The larger volume of 1.3 and 1.9 mm rotors (2.5 and 13.1 µL, respectively) represents a significant sensitivity advantage over a 0.7 mm rotor (0.59 µL). Le Marchand et al. (2022) estimated a factor of ca. 2.5 lower detection sensitivity for a 0.7 mm probe, due to the smaller rotor (Fig. 9 of Le Marchand et al., 2022). An experimental study compared 1.3 and 1.9 mm rotors and reported a sensitivity gain of the 1.9 mm rotor by a factor of ca. 1.2–1.9 (Nieuwkoop et al., 2015). The sensitivity ultimately also depends on the losses during the pulse sequence and the relaxation properties. Given the apparent decay rate constants in H2O/M9/lysate-deuterated and protonated samples (Fig. 3a, b), the approach proposed here likely provides superior sensitivity also for complex experiments with many transfer steps. Lastly, while protonated samples are cheaper than the ones we used here, 0.7 mm rotors are more expensive (ca. EUR 3800) than 1.3 mm (ca. EUR 2600) or 1.9 mm rotors (EUR 2000), and in our hands the process of opening and reusing 0.7 mm rotors is more prone to damage, in particular of caps (a cap also costs ca. EUR 250–300). Again, these prices are to be taken with a grain of salt, but they illustrate that currently the proposed H2O/M9/lysate approach is also economically advantageous.
Our approach can of course also be used with a H2O / D2O mixture, which will result in even higher deuteration levels. For example, using 50 % D2O in the growth medium will result in 50 % protonation of the non-exchangeable sites and 100 % for the exchangeable sites (provided the final sample is in H2O). Using this approach enables the detection of all amide sites without lengthy exploration of refolding protocols with unsure outcomes. In new projects in our group, we apply this approach right from the start whenever the involved protein is likely to have non-exchangeable amides, thereby saving precious time and ensuring high-quality samples.
4.1 Protein samples
Pyrococcus horikoshii TET2 production was achieved by overexpressing pET-41c plasmid in E. coli BL21(DE3) competent cells (Novagen). The plasmid is available at AddGene (deposition number 182428). Culture media based on M9 minimal media were used for the different labeling schemes: (i) u-[2H,13C,15N]-labeled (perdeuterated) samples were expressed in M9 minimal media, 99.8 % D2O, 15NH4Cl as sole nitrogen source, deuterated, and 13C6-labeled glucose; (ii) H2O/M9/lysate-deuterated samples were expressed in H2O M9 minimal media, supplemented with u-[2H,13C,15N]-labeled algal lysates (2 g L−1 of culture unless specified otherwise; Sigma-Aldrich) and 2H,13C D-glucose as an additional carbon and energy source; (iii) u-[1H,13C,15N]-labeled (protonated) samples were expressed in H2O M9 minimal media, integrating 15NH4Cl and protonated 13C6 D-glucose. Cells were grown at 37 °C until optical density (OD, 600 nm) reached ca. 0.6–0.8. Protein expression was induced using isopropyl-β-D-1-thiogalactopyranoside (IPTG; 1 mM). Cells were harvested by centrifugation, and pellets were resuspended in Lysis buffer T (Table S2) and disrupted using a Microfluidizer (M-110 P, Microfluidics). Cell lysates were heated to 85 °C for 15 min and subsequently centrifuged (17 500 rcf (relative centrifugal force), 1 h, 4 °C). The supernatant was dialyzed overnight against Dialysis buffer at room temperature and recentrifuged as in the previous step. Protein purification was conducted using a Resource Q column (GE Healthcare), eluting TET2 using a linear gradient of Elution buffer over 10 column volumes. Fractions were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (12.5 % polyacrylamide), and TET2 was identified by its monomeric molecular weight (39 kDa) and concentrated using an Amicon® 30 kDa concentrator (Millipore). The concentrated solution was loaded onto a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated with Dialysis buffer. Samples for MAS NMR measurements were prepared as described earlier (Gauto et al., 2019a), by concentrating TET2 to 10 mg mL−1 in NMR buffer. The solution was then mixed (1:1 vol/vol) with 2-methyl-2,4-pentanediol (MPD). Protein precipitates were packed into 0.7/1.3/1.6/3.2 mm MAS rotors by ultracentrifugation (50 000 g, at least 1 h).
I. islandicus malate dehydrogenase was produced by overexpression of the pET-21a(+) plasmid in E. coli BL21(DE3) competent cells (Novagen). M9 minimal-based media was used as for TET2; H2O/M9/lysate-deuterated cultures were supplemented with 1, 2, or 4 g L−1 of culture of u-[2H,13C,15N]-labeled ISOGRO®, as well as the standard 2 g L−1 of 2H,13C D-glucose and 1 g L−1 15NH4Cl. Cells were grown at 37 °C until OD (600 nm) reached ca. 0.6–0.8. Protein expression was induced using IPTG (1 mM). Cells were harvested by centrifugation after 3–5 h of expression, and pellets were resuspended in Lysis buffer M. Lysis was performed by sonication, using a Q700 ultrasonic processor (Qsonica), at 40 % amplitude for a total operating time of 6 min. Cell lysates were heated to 70 °C for 20 min and subsequently centrifuged (40 000 rcf, 1 h, 4 °C). The supernatant was filtered, and protein purification was conducted using a Resource Q column (GE Healthcare) equilibrated in Buffer A, eluting the protein using a linear gradient of Buffer B over 10 column volumes. Fractions were analyzed by SDS-PAGE (12.5 % polyacrylamide), and the protein was identified by its monomeric molecular weight (33.55 kDa) and concentrated using an Amicon® 10 kDa concentrator (Millipore). The concentrated solution was therefore loaded onto a HiLoad 26/600 Superdex 200 PG column (Sigma-Aldrich) equilibrated with Buffer A. MAS rotors were filled for NMR measurements by ultracentrifugation (68 300 rcf, over night).
Bacteriophage T5 tail tube protein pb6 modified in C-terminal with Tobacco Etch Virus (TEV) protease cleaving site and hexahistidine-tag was produced by overexpression in E. coli BL21(DE3) (Singles™ competent cells, Merck) of a pb6-pLIM13 plasmid. Cells were grown either (i) from deuterated preculture in D2O-based minimal buffer (M9) or (ii) with home-made u-[2H,13C,15N] cell lysate from a deuterated bacterial culture added at a concentration of 2 g L−1 to a H2O/M9 culture medium. 13C-labeled glucose (2 g L−1) and 15NH4 (1 g L−1) were used in the medium in both cases. The purification protocol is reported in Arnaud et al. (2017). Briefly, cell lysis was performed by six passages through a Microfluidizer (M-110 P, Microfluidics). Cell lysates were centrifuged for 30 min at 3000 rpm (rotations per minute type 45 Ti rotor) and 4 °C, and pellets were resuspended and incubated for 1 h at 37 °C in Resuspension buffer P. They were then applied to a sucrose cushion gradient (50 %, 40 %, 35 %, and 35 % sucrose cushion, Sucrose buffer) and spun for 30 min at 30 000 rpm (SW41 rotor, Beckman Coulter) and 4 °C. The obtained pellet was energetically resuspended in Buffer P and pelleted/resuspended several times by low-speed centrifugation to remove sucrose contamination.
Ubiquitin samples, prepared to estimate the labeling amount, were expressed in E. coli BL21(DE3) cells, transformed with a pET-21b plasmid carrying the human ubiquitin gene. For the production of perdeuterated ubiquitin, transformants were adapted progressively in four stages over 48 h to M9 / D2O media containing 1 g L−1 15ND4Cl and 2 g L−1 2H,13C D-glucose as the sole nitrogen and carbon sources. In the final culture, the bacteria were grown at 37 ∘C. When the optical density at 600 nm (OD600) reached ca. 0.8–0.9, protein expression was induced by addition of IPTG to a final concentration of 1 mM, and cells were incubated for another 3 h at 37 °C. For the ubiquitin samples prepared with cell lysate (either ISOGRO® or in-house cell lysate; see below) we used either the same adaptation protocol and a D2O-based preculture or a shortened protocol without adaption state and with a H2O-based preculture. The D2O preculture does not have an effect on the final labeling (Fig. 2b). In all cases, cells were harvested by centrifugation, resuspended in 20 mL of Lysis buffer U, and lysed by sonication. The lysate was centrifuged for 30 min at 46 000 g (JA25-50 Beckman rotor), and the supernatant was dialyzed against two times 300 mL of buffer U. After dialysis, the sample was centrifuged for 30 min at 46 000 g and loaded on a 40 mL Q-Sepharose column. Ubiquitin was recovered in the flow-through fractions, which were subsequently concentrated and injected on a HiLoad 16/60 Superdex 75 column equilibrated with 1 column volume of buffer U. The buffer was exchanged to pH 6.5 for solution-state NMR.
4.2 NMR spectroscopy
MAS NMR spectra of the proteins TET2 (except those of Fig. 3) and pb6 were acquired on a Bruker Avance 3 HD (600 MHz) equipped with a 1.3 mm probe tuned to 1H, 13C, 15N with an auxiliary coil tuned to 2H. The effective sample temperature of MAS NMR experiments was kept at ca. 28 °C, measured from the bulk water frequency, using the relationship T[°C]=255–90 ⋅ δH20, where δH20 is the bulk water (Böckmann et al., 2009) frequency in ppm. The chemical shift was referenced with respect to the signal of MPD at 4.1 ppm, which is not significantly dependent on temperature (Fig. S1 of Gauto et al., 2019b). Additional spectra of TET2 at 100 kHz MAS frequency (Fig. 6) were recorded on a Bruker Avance 3 HD (950 MHz) spectrometer equipped with a 0.7 mm probe tuned to 1H, 13C, and 15N. Experiments for T2′ measurements (Fig. 3) were recorded on a Bruker NEO spectrometer (700 MHz) equipped with a 0.7 mm HCN probe (Fig. 3). Experiments with MalDH were recorded on Bruker NEO spectrometers operating at 600 or 700 MHz as specified in Fig. 4d, e, using 1.9 mm HXY probes tuned to 1H, 13C, and 15N.
All experiments reported herein (2D hNH, 2D hCH, 3D hCANH, 3D hCONH, 4D HACANH) were recorded with pulse sequences available in the NMRlib library (Vallet et al., 2020). All transfers in these experiments used cross-polarization transfer steps. The 4D HCANH (Fig. 6a) used the following CP parameters: 40 kHz (13C) and 92 kHz (1H, linear ramp 90–100) for the H–CA transfer (4 ms), 12.5 kHz (13C) and 42 kHz (15N, linear ramp 70–100) for the CA–N transfer (8 ms), and 40 kHz (15N) and ca. 90 kHz (1H, linear ramp 90–100) for the N–H transfer (1 ms). The indirect dimensions were as follows: CA 34 ppm, 88 points; N 32 ppm, 120 points; and HA 12 ppm, 58 points. The recycle delay was set to 0.65 s, and the total duration was ca. 6 d. Similar CP parameters were used for hCONH, hCANH, hNH and hCH experiments recorded at 55 kHz. Typical experimental times for the 3D experiments were 2 d.
The hNCAHA experiment performed at 100 kHz (950 MHz) used the following CP parameters. H–N CP at 22 kHz (15N) and 122 kHz (1H, ramp 90–100; 1 ms), N–CA 38 kHz (15N, ramp 90–100, 4 ms) and 70 kHz (13C), and CA–HA CP at 30 kHz (13C) and 125 kHz (1H, ramp 90–100, 1 ms); the total experimental time was 2 d. The CP setting used for the hCH experiment was the same as the CA–HA setting above.
In the following, note that the italic values were used to acquire the perdeuterated sample spectrum, while the non-italic values were used for the H2O/M9/lysate spectrum. The hCANH experiment performed at 38 kHz for MalDH (600/700 MHz) used the following CP parameters. H–N CP at 24/48 kHz (15N) and 63/85 kHz (1H, ramp 90–100; 1 ms) H–CA CP at 23/50 kHz (13C) and 63/82 kHz (1H, ramp 90–100; 4 ms), N–CA 28/30 kHz (15N, ramp 90–100; 10 ms) and 8/7 kHz (13C). The experimental time was 2 d.
Solution-state NMR of ubiquitin was performed on a 700 MHz Bruker Avance 3 HD spectrometer equipped with a cryo-probe to quantify the amount of deuteration. 13C-HSQC experiments were recorded on samples of ca. 0.3–0.5 mM concentration with a 13C spectral width of 70 ppm (512 complex points) using a 3 s recycle delay. The intensity was normalized by the sample concentration (confirmed by 1D 1H spectral intensity), and the deuteration level was obtained by comparing the peak intensities of aliphatic signals with those of the fully protonated sample. The results for the Hα sites were confirmed using an alternative method based on HNcoCA experiments omitting 1H decoupling, as done by Löhr et al. (2003).
For all NMR spectra, spectral inspection, manual peak-picking, and chemical shifts assignment were performed using the CCPNMR software (Vranken et al., 2005). Routines for fits of coherence decays (Fig. S1 in the Supplement) were written in Python.
4.3 Mass spectrometry
The deuteration level of MalDH produced with different culture-media compositions (Fig. 1a) was determined by intact mass spectrometry at the MS facility of the Max Perutz Labs. Briefly, samples were diluted to 20 ng µL−1 in H2O, and 30–40 ng was loaded on a XBridge Protein BEH C4 Column, 300 Å, 2.5 µm, 2.1 mm ×150 mm (Waters), using a Dionex Ultimate 3000 high-performance liquid chromatography (HPLC) system (Thermo Scientific). The proteins were eluted with an acetonitrile gradient from 12 % to 72 % in 0.1 % formic acid at a flow rate of 250 µL min−1. Mass spectra were recorded in the resolution mode on a Synapt G2-Si mass spectrometer equipped with a ZSpray ESI source (Waters). Glu[1]-Fibrinopeptide B (Glu-Fib) was used as a lock mass, and spectra were corrected on the fly. Data were analyzed in MassLynx version 4.1 using the MaxEnt 1 process to reconstruct the uncharged average protein mass.
4.4 Preparation of in-house cell lysate
We prepared a substitute for the commercial algal medium by using unused parts from cultures prepared under perdeuteration conditions, i.e., from proteins usually considered contaminants. Standard perdeuteration conditions were used for the cultures (D2O, 2 g L−1 2H,13C glucose, 1 g L−1 15N ammonium chloride). We prepared such cell lysates from two independent cultures. The proteins produced in these cultures were either TET2 or ClpP (as reported in Felix et al., 2019). In both cases, the proteins are expressed in the soluble fraction (rather than in the pellet of the membrane/inclusion bodies). In one experiment, performed with ClpP, the protein of interest was purified by Ni-affinity chromatography. All wash fractions from this affinity chromatography step (prior to eluting ClpP) with significant amount of protein were pooled and dialyzed against water to eliminate the imidazol contained in the wash steps. The solution containing the contaminant proteins was then acidified with 1 M phosphoric acid (1 M final) and left at 80 °C in an incubation oven for 5 d to allow for acid-catalyzed peptide-bond hydrolysis. The rationale for using phosphoric acid is that after neutralization the sample contains phosphate, a buffer component used in M9 media. Thereafter, it was neutralized with NaOH, cleared by centrifugation, and lyophilized.
The second approach, performed with ClpP and TET2, used the debris after lysis of the cells and centrifugation; rather than using the soluble fraction that was taken above, the insoluble fraction was subjected to the same acid hydrolysis treatment described above. The amino acid composition from all three samples was very similar.
4.5 Amino acid analysis
For the determination of the amino acid composition of the in-house lysate, the lyophilized powder of the samples described in the previous section, doped with a precisely known amount of norleucine, was dried and then hydrolyzed for 24 h at 110 °C in 6 N HCl containing 1 % () phenol. Samples were then dried and resuspended in analysis buffer and injected into an ion-exchange column of a Biochrom 30 amino acid analyzer, using measurements of the optical density at 570 and 440 nm. Quantification was performed with respect to the internal norleucine standard.
All pulse sequences are available in the NMRlib library (Vallet et al., 2020) and from the authors. Analysis scripts for coherence-decay experiments and linewidth analyses are available from the authors. The new resonance assignments were submitted to the BioMagResBank (https://doi.org/10.13018/BMR52400, Napoli and Schanda, 2024). Other data are available from the authors upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/mr-5-33-2024-supplement.
FN prepared all MalDH samples and TET samples for T2′ measurements, recorded and analyzed all T2′ and linewidth data, recorded all multidimensional experiments of MalDH, and performed TET2 resonance assignment and TALOS analyses. JYG prepared ubiquitin samples and analyzed the labeling pattern and a TET2 sample. C-AA and CB prepared pb6 samples. PM and HF prepared cell lysates from bacterial cultures. PS designed the study and recorded NMR experiments. FN and PS prepared all figures and wrote the article.
At least one of the (co-)authors is a member of the editorial board of Magnetic Resonance. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We thank Dominique Madern (IBS Grenoble) for providing the plasmid for MalDH and feedback on the article, Alicia Vallet for excellent support at the Grenoble NMR facility, and Petra Rovo and Margarita Valhondo at the IST Austria NMR Service Unit. We thank Dorothea Anrather in the mass spectrometry facility of Max Perutz Labs for the mass spectrometry analysis using the instruments of the Vienna BioCenter Core Facilities (VBCF). We are grateful to Jean-Pierre Andrieu (Plateforme Seq3A, IBS Grenoble) for the analysis of the amino acid composition of the in-house-prepared lysates. We are grateful to Rasmus Linser (Technical University Dortmund) for sharing a paper draft describing a similar study. This work was supported by the Austrian Science Fund (FWF; project number I5812-B). We thank Tobias Schubeis (Lyon) and the reviewers for constructive input.
This research has been supported by the Austrian Science Fund (grant no. I5812-B). Part of this work used the platforms of the Grenoble Instruct-ERIC center (ISBG; UAR 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for 40 Structural Biology (PSB), supported by FRISBI (ANR-10-INBS-0005-02) and GRAL, financed within the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). IBS acknowledges integration into the Interdisciplinary Research Institute of Grenoble (IRIG, 45 CEA). Charles-Adrien Arnaud was funded by GRAL.
This paper was edited by Beat Meier and reviewed by two anonymous referees.
Agarwal, V., Penzel, S., Szekely, K., Cadalbert, R., Testori, E., Oss, A., Past, J., Samoson, A., Ernst, M., Böckmann, A., and Meier, B. H.: De novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy, Angew. Chem. Int. Edit., 53, 12253–12256, 2014. a
Anderson, E.: Growth requirements of virus-resistant mutants of Escherichia coli strain “B”, P. Natl. Acad. Sci. USA, 32, 120–128, 1946. a
Andreas, L. B., Stanek, J., Le Marchand, T., Bertarello, A., Paepe, D. C.-D., Lalli, D., Krejčíková, M., Doyen, C., Öster, C., Knott, B., Wegner, S., Engelke, F., Felli, I. C., Pierattelli, R., Dixon, N. E., Emsley, L., Herrmann, T., and Pintacuda, G.: Protein residue linking in a single spectrum for magic-angle spinning NMR assignment, J. Biomol. NMR, 62, 253–261, 2015. a
Andreas, L. B., Jaudzems, K., Stanek, J., Lalli, D., Bertarello, A., Le Marchand, T., Cala-De Paepe, D., Kotelovica, S., Akopjana, I., Knott, B., Wegner, S., Engelke, F., Lesage, A., Emsley, L., Tars, K., Herrmann, T., and Pintacuda, G.: Structure of fully protonated proteins by proton-detected magic-angle spinning NMR, P. Natl. Acad. Sci. USA, 113, 9187–9192, 2016. a
Arnaud, C.-A., Effantin, G., Vivès, C., Engilberge, S., Bacia, M., Boulanger, P., Girard, E., Schoehn, G., and Breyton, C.: Bacteriophage T5 tail tube structure suggests a trigger mechanism for Siphoviridae DNA ejection, Nat. Commun., 8, 1953, https://doi.org/10.1038/s41467-017-02049-3, 2017. a
Asami, S., Schmieder, P., and Reif, B.: High resolution 1H-detected solid-state NMR spectroscopy of protein aliphatic resonances: Access to tertiary structure information, J. Am. Chem. Soc., 132, 15133–15135, 2010. a, b
Asami, S., Szekely, K., Schanda, P., Meier, B. H., and Reif, B.: Optimal degree of protonation for 1H detection of aliphatic sites in randomly deuterated proteins as a function of the MAS frequency, J. Biomol. NMR, 54, 155–168, 2012. a, b
Aucharova, H., Klein, A., Medina, S., Söldner, B., Vasa, S. K., and Linser, R.: Protein deuteration via algal amino acids to overcome proton back-exchange for fast- MAS solid-state NMR of large proteins, Chem. Commun., 60, 3083–3086, https://doi.org/10.1039/D4CC00213J, 2024. a, b
Barbet-Massin, E., Pell, A. J., Retel, J. S., Andreas, L. B., Jaudzems, K., Franks, W. T., Nieuwkoop, A. J., Hiller, M., Higman, V., Guerry, P., Bertarello, A., Knight, M. J., Felletti, M., Le Marchand, T., Kotelovica, S., Akopjana, I., Tars, K., Stoppini, M., Bellotti, V., Bolognesi, M., Ricagno, S., Chou, J. J., Griffin, R. G., Oschkinat, H., Lesage, A., Emsley, L., Herrmann, T., and Pintacuda, G.: Rapid proton-detected NMR assignment for proteins with fast magic angle spinning, J. Am. Chem. Soc., 136, 12489–12497, 2014. a
Böckmann, A., Gardiennet, C., Verel, R., Hunkeler, A., Loquet, A., Pintacuda, G., Emsley, L., Meier, B. H., and Lesage, A.: Characterization of different water pools in solid-state NMR protein samples, J. Biomol. NMR, 45, 319–327, 2009. a
Bonaccorsi, M., Le Marchand, T., and Pintacuda, G.: Protein structural dynamics by magic-angle spinning NMR, Curr. Opin. Struc. Biol., 70, 34–43, 2021. a
Bougault, C., Ayala, I., Vollmer, W., Simorre, J.-P., and Schanda, P.: Studying intact bacterial peptidoglycan by proton-detected NMR spectroscopy at 100 kHz MAS frequency, J. Struct. Biol., 206, 66–72, 2019. a
Cala-De Paepe, D., Stanek, J., Jaudzems, K., Tars, K., Andreas, L. B., and Pintacuda, G.: Is protein deuteration beneficial for proton detected solid-state NMR at and above 100 kHz magic-angle spinning?, Solid State Nucl. Magn. Reson., 87, 126–136, 2017. a, b
Chevelkov, V., Rehbein, K., Diehl, A., and Reif, B.: Ultrahigh resolution in proton solid-state NMR spectroscopy at high levels of deuteration, Angew. Chem. Int. Edit., 45, 3878–3881, 2006. a
Chevelkov, V., Fink, U., and Reif, B.: Quantitative analysis of backbone motion in proteins using MAS solid-state NMR spectroscopy, J. Biomol. NMR, 45, 197–206, 2009. a
Felix, J., Weinhäupl, K., Chipot, C., Dehez, F., Hessel, A., Gauto, D. F., Morlot, C., Abian, O., Gutsche, I., Velazquez-Campoy, A., Schanda, P., and Fraga, H.: Mechanism of the allosteric activation of the ClpP protease machinery by substrates and active-site inhibitors, Sci. Adv., 5, eaaw3818, https://doi.org/10.1126/sciadv.aaw3818 , 2019. a
Fraga, H., Arnaud, C.-A., Gauto, D. F., Audin, M., Kurauskas, V., Macek, P., Krichel, C., Guan, J.-Y., Boisbouvier, J., Sprangers, R., Breyton, C., and Schanda, P.: Solid-State NMR H-N-(C)-H and H-N-C-C 3D/4D Correlation Experiments for Resonance Assignment of Large Proteins, Chem. Phys. Chem., 18, 2697–2703, 2017. a
Gardner, K. H. and Kay, L. E.: The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins, Annu. Rev. Bioph. Biom., 27, 357–406, 1998. a
Gardner, K. H., Zhang, X., Gehring, K., and Kay, L. E.: Solution NMR Studies of a 42 kDa Escherichia Coli Maltose Binding Protein/β-Cyclodextrin Complex: Chemical Shift Assignments and Analysis, J. Am. Chem. Soc., 120, 11738–11748, 1998. a
Gauto, D. F., Estrozi, L. F., Schwieters, C. D., Effantin, G., Macek, P., Sounier, R., Sivertsen, A. C., Schmidt, E., Kerfah, R., Mas, G., Colletier, J.-P., Güntert, P., Favier, A., Schoehn, G., Schanda, P., and Boisbouvier, J.: Integrated NMR and cryo-EM atomic-resolution structure determination of a half-megadalton enzyme complex, Nat. Commun., 10, 1–12, 2019a. a, b, c, d, e, f
Gauto, D. F., Macek, P., Barducci, A., Fraga, H., Hessel, A., Terauchi, T., Gajan, D., Miyanoiri, Y., Boisbouvier, J., Lichtenecker, R., Kainosho, M., and Schanda, P.: Aromatic Ring Dynamics, Thermal Activation, and Transient Conformations of a 468 kDa Enzyme by Specific 1H–13C Labeling and Fast Magic-Angle Spinning NMR, J. Am. Chem. Soc., 141, 11183–11195, 2019b. a
Gauto, D. F., Macek, P., Malinverni, D., Fraga, H., Paloni, M., Sučec, I., Hessel, A., Bustamante, J. P., Barducci, A., and Schanda, P.: Functional control of a 0.5 MDa TET aminopeptidase by a flexible loop revealed by MAS NMR, Nat. Commun., 13, 1927, https://doi.org/10.1038/s41467-022-29423-0, 2022. a
Good, D. B., Wang, S., Ward, M. E., Struppe, J., Brown, L. S., Lewandowski, J. R., and Ladizhansky, V.: Conformational dynamics of a seven transmembrane helical protein Anabaena Sensory Rhodopsin probed by solid-state NMR, J. Am. Chem. Soc., 136, 2833–2842, 2014. a
Huber, M., Hiller, S., Schanda, P., Ernst, M., Böckmann, A., Verel, R., and Meier, B. H.: A Proton-Detected 4D Solid-State NMR Experiment for Protein Structure Determination, Chem. Phys. Chem., 12, 915–918, 2011. a
Imbert, L., Lenoir-Capello, R., Crublet, E., Vallet, A., Awad, R., Ayala, I., Juillan-Binard, C., Mayerhofer, H., Kerfah, R., Gans, P., Miclet, E., and Boisbouvier, J.: In Vitro Production of Perdeuterated Proteins in H2O for Biomolecular NMR Studies, in: Meth. Mol. Biol., vol. 2199, 127–149, Humana, 2021. a
Jain, M. G., Lalli, D., Stanek, J., Gowda, C., Prakash, S., Schwarzer, T. S., Schubeis, T., Castiglione, K., Andreas, L. B., Madhu, P. K., Pintacuda, G., and Agarwal, V.: Selective 1H-1H Distance Restraints in Fully Protonated Proteins by Very Fast Magic-Angle Spinning Solid-State NMR, J. Phys. Chem. Lett., 8, 2399–2405, 2017. a
Klein, A., Rovó, P., Sakhrani, V. V., Wang, Y., Holmes, J. B., Liu, V., Skowronek, P., Kukuk, L., Vasa, S. K., Güntert, P., Mueller, L. J., and Linser, R.: Atomic-resolution chemical characterization of (2x) 72-kDa tryptophan synthase via four-and five-dimensional 1H-detected solid-state NMR, P. Natl. Acad. Sci. USA, 119, e2114690119, https://doi.org/10.1073/pnas.2114690119, 2022. a
Knight, M. J., Pell, A. J., Bertini, I., Felli, I. C., Gonnelli, L., Pierattelli, R., Herrmann, T., Emsley, L., and Pintacuda, G.: Structure and backbone dynamics of a microcrystalline metalloprotein by solid-state NMR, P. Natl. Acad. Sci. USA, 109, 11095–11100, 2012. a, b
Lamley, J. M., Iuga, D., Öster, C., Sass, H.-J., Rogowski, M., Oss, A., Past, J., Reinhold, A., Grzesiek, S., Samoson, A., and Lewandowski, J. R.: Solid-state NMR of a protein in a precipitated complex with a full-length antibody, J. Am. Chem. Soc., 136, 16800–16806, 2014. a
Le Marchand, T., Schubeis, T., Bonaccorsi, M., Paluch, P., Lalli, D., Pell, A. J., Andreas, L. B., Jaudzems, K., Stanek, J., and Pintacuda, G.: 1H-detected biomolecular NMR under fast magic-angle spinning, Chem. Rev., 122, 9943–10018, 2022. a, b, c, d, e, f, g, h
Lewandowski, J. R.: Advances in Solid-State Relaxation Methodology for Probing Site-Specific Protein Dynamics, Acc. Chem. Res., 46, 2018–2027, 2013. a
Lewandowski, J. R., Dumez, J. N., Akbey, U., Lange, S., Emsley, L., and Oschkinat, H.: Enhanced Resolution and Coherence Lifetimes in the Solid-state NMR Spectroscopy of Perdeuterated Proteins under Ultrafast Magic-angle Spinning, J. Phys. Chem. Lett., 2, 2205–2211, 2011. a
Linser, R., Bardiaux, B., Higman, V., Fink, U., and Reif, B.: Structure Calculation from Unambiguous Long-Range Amide and Methyl 1 H− 1 H Distance Restraints for a Microcrystalline Protein with MAS Solid-State NMR Spectroscopy, J. Am. Chem. Soc., 133, 5905–5912, 2011a. a
Linser, R., Bardiaux, B., Higman, V., Fink, U., and Reif, B.: Structure Calculation from Unambiguous Long-Range Amide and Methyl 1H-1H Distance Restraints for a Microcrystalline Protein with MAS Solid-State NMR Spectroscopy, J. Am. Chem. Soc., 133, 5905–5912, 2011b. a
Linser, R., Sarkar, R., Krushelnitzky, A., Mainz, A., and Reif, B.: Dynamics in the solid-state: perspectives for the investigation of amyloid aggregates, membrane proteins and soluble protein complexes, J. Biomol. NMR, 59, 1–14, 2014. a
Löhr, F., Katsemi, V., Hartleib, J., Günther, U., and Rüterjans, H.: A strategy to obtain backbone resonance assignments of deuterated proteins in the presence of incomplete amide 2H/1H back-exchange, J. Biomol. NMR, 25, 291–311, 2003. a, b, c, d, e, f, g
Medeiros-Silva, J., Mance, D., Daniëls, M., Jekhmane, S., Houben, K., Baldus, M., and Weingarth, M.: 1H-Detected Solid-State NMR Studies of Water-Inaccessible Proteins In Vitro and In Situ, Angew. Chem. Int. Edit., 55, 13606–13610, 2016. a, b, c, d, e, f, g
Najbauer, E. E., Tekwani Movellan, K., Giller, K., Benz, R., Becker, S., Griesinger, C., and Andreas, L. B.: Structure and gating behavior of the human integral membrane protein VDAC1 in a lipid bilayer, J. Am. Chem. Soc., 144, 2953–2967, 2022. a
Napoli, F. and Schanda, P.: Amide and α-hydrogens extension to the Solid-state NMR assignment of P. horikoshii TET2, BioMagResBank Entry 52400 [data set], https://doi.org/10.13018/BMR52400, 2024. a
Napoli, F., Becker, L. M., and Schanda, P.: Protein dynamics detected by magic-angle spinning relaxation dispersion NMR, Curr. Opin. Struct. Biol., 82, 102660, https://doi.org/10.1016/j.sbi.2023.102660, 2023. a
Nieuwkoop, A. J., Franks, W. T., Rehbein, K., Diehl, A., Akbey, U., Engelke, F., Emsley, L., Pintacuda, G., and Oschkinat, H.: Sensitivity and resolution of proton detected spectra of a deuterated protein at 40 and 60 kHz magic-angle-spinning, J. Biomol. NMR, 61, 161–171, 2015. a
O'Brien, E. S., Lin, D. W., Fuglestad, B., Stetz, M. A., Gosse, T., Tommos, C., and Wand, A. J.: Improving yields of deuterated, methyl labeled protein by growing in H2O, J. Biomol. NMR, 71, 263–273, 2018. a, b, c
Pervushin, K., Riek, R., Wider, G., and Wüthrich, K.: Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution, P. Natl. Acad. Sci. USA, 94, 12366–12371, 1997. a
Pervushin, K., Riek, R., Wider, G., and Wüthrich, K.: Transverse relaxation-optimized spectroscopy (TROSY) for NMR studies of aromatic spin systems in 13C-labeled proteins, J. Am. Chem. Soc., 120, 6394–6400, 1998. a
Reif, B.: Deuteration for high-resolution detection of protons in protein magic angle spinning (MAS) solid-state NMR, Chem. Rev., 122, 10019–10035, 2021. a
Retel, J. S., Nieuwkoop, A. J., Hiller, M., Higman, V. A., Barbet-Massin, E., Stanek, J., Andreas, L. B., Franks, W. T., Van Rossum, B.-J., Vinothkumar, K. R., Handel, L., de Palma, G. G., Bardiaux, B., Pintacuda, G., Emsley, L., Kühlbrandt, and Oschkinat, H.: Structure of outer membrane protein G in lipid bilayers, Nat. Commun., 8, 2073, https://doi.org/10.1038/s41467-017-02228-2, 2017. a
Schmidt, E. and Güntert, P.: A New Algorithm for Reliable and General NMR Resonance Assignment, J. Am. Chem. Soc., 134, 12817–12829, 2012. a
Schubeis, T., Le Marchand, T., Daday, C., Kopec, W., Tekwani Movellan, K., Stanek, J., Schwarzer, T. S., Castiglione, K., de Groot, B. L., Pintacuda, G., and Andreas L. B.: A β-barrel for oil transport through lipid membranes: Dynamic NMR structures of AlkL, P. Natl. Acad. Sci. USA, 117, 21014–21021, 2020. a
Shaka, A. J., Keeler, J., and Freeman, R.: Evaluation of a new broad-band decoupling sequence – WALTZ-16, J. Magn. Reson., 53, 313–340, 1983. a
Shcherbakov, A. A., Mandala, V. S., and Hong, M.: High-Sensitivity Detection of Nanometer 1H-19F Distances for Protein Structure Determination by 1H-Detected Fast MAS NMR, J. Phys. Chem. B, 123, 4387–4391, 2019. a
Shen, Y. and Bax, A.: Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks, J. Biomol. NMR, 56, 227–241, 2013. a
Singh, H., Vasa, S. K., Jangra, H., Rovó, P., Päslack, C., Das, C. K., Zipse, H., Schäfer, L. V., and Linser, R.: Fast Microsecond Dynamics of the Protein–Water Network in the Active Site of Human Carbonic Anhydrase II Studied by Solid-State NMR Spectroscopy, J. Am. Chem. Soc., 141, 19276–19288, 2019. a
Stanek, J., Schubeis, T., Paluch, P., Güntert, P., Andreas, L. B., and Pintacuda, G.: Automated Backbone NMR Resonance Assignment of Large Proteins Using Redundant Linking from a Single Simultaneous Acquisition, J. Am. Chem. Soc. Soc., 142, 5793–5799, 2020. a
Tekwani, K., Eszter, M., Supriya, E. N., Michele, P., Karin, S., Stefan, G., and Andreas, L. B.: Alpha protons as NMR probes in deuterated proteins, J. Biomol. NMR, 73, 81–91, 2019. a
Tugarinov, V., Hwang, P. M., Ollerenshaw, J. E., and Kay, L. E.: Cross-correlated relaxation enhanced 1H-13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes, J. Am. Chem. Soc., 125, 10420–10428, 2003. a
Vallet, A., Favier, A., Brutscher, B., and Schanda, P.: ssNMRlib: a comprehensive library and tool box for acquisition of solid-state nuclear magnetic resonance experiments on Bruker spectrometers, Magn. Reson., 1, 331–345, https://doi.org/10.5194/mr-1-331-2020, 2020. a, b
Vasa, S. K., Rovó, P., and Linser, R.: Protons as versatile reporters in solid-state NMR spectroscopy, Acc. Chem. Res., 51, 1386–1395, 2018. a
Vranken, W. F., Boucher, W., Stevens, T. J., Fogh, R. H., Pajon, A., Llinas, M., Ulrich, E. L., Markley, J. L., Ionides, J., and Laue, E. D.: The CCPN data model for NMR spectroscopy: development of a software pipeline, Proteins, 59, 687–96, 2005. a
Xiang, S., Grohe, K., Rovó, P., Vasa, S. K., Giller, K., Becker, S., and Linser, R.: Sequential backbone assignment based on dipolar amide-to-amide correlation experiments, J. Biomol. NMR, 62, 303–311, https://doi.org/10.1007/s10858-015-9945-4, 2015. a
Xuncheng, S., Loh, C.-T., Ruhu, Q., and Otting, G.: Suppression of isotope scrambling in cell-free protein synthesis by broadband inhibition of PLP enymes for selective 15N-labelling and production of perdeuterated proteins in H2O, J. Biomol. NMR, 50, 35–42, https://doi.org/10.1007/s10858-011-9477-5, 2011. a
Zhou, D. H., Shea, J. J., Nieuwkoop, A. J., Franks, W. T., Wylie, B. J., Mullen, C., Sandoz, D., and Rienstra, C. M.: Solid-State Protein-Structure Determination with Proton-Detected Triple-Resonance 3D Magic-Angle-Spinning NMR Spectroscopy, Angew. Chem. Int. Ed., 119, 8532–8535, 2007. a, b