the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Nov 2020
| 13 Nov 2020
Analysis of the electronic structure of the primary electron donor of photosystem I of Spirodela oligorrhiza by photochemically induced dynamic nuclear polarization (photo-CIDNP) solid-state nuclear magnetic resonance (NMR)
Geertje J. Janssen
Patrick Eschenbach
Patrick Kurle
Bela E. Bode
Johannes Neugebauer
Huub J. M. de Groot
Jörg Matysik
Alia Alia
The electron donor in photosystem I (PSI), the chlorophyll dimer P700, is studied by photochemically induced dynamic nuclear polarization (photo-CIDNP) magic angle spinning (MAS) nuclear magnetic resonance (NMR) on selectively 13C and uniformly 15N labeled PSI core preparations (PSI-100) obtained from the aquatic plant duckweed (Spirodela oligorrhiza). Light-induced signals originate from the isotope-labeled nuclei of the cofactors involved in the spin-correlated radical pair forming upon light excitation. Signals are assigned to the two donor cofactors (Chl a and Chl a') and the two acceptor cofactors (both Chl a). Light-induced signals originating from both donor and acceptor cofactors demonstrate that electron transfer occurs through both branches of cofactors in the pseudo-C2 symmetric reaction center (RC). The experimental results supported by quantum chemical calculations indicate that this functional symmetry occurs in PSI despite similarly sized chemical shift differences between the cofactors of PSI and the functionally asymmetric special pair donor of the bacterial RC of Rhodobacter sphaeroides. This contributes to converging evidence that local differences in time-averaged electronic ground-state properties, over the donor are of little importance for the functional symmetry breaking across photosynthetic RC species.
- Article
(2663 KB) - Full-text XML
-
Supplement
(2181 KB) - BibTeX
- EndNote





In the process of oxygenic photosynthesis, electrons flow from photosystem II (PSII) to photosystem I (PSI); the nomenclature, however, follows the order of their discovery over time (Emerson and Chalmers, 1958; Govindjee and Rabinowitch, 1960). The X-ray structure of PSI from the prokaryotic system of cyanobacteria Synechococcus (S.) elongatus has been solved at 2.5 Å resolution as a trimeric supercomplex (Jordan et al., 2001). In the eukaryotic plant system of Pisum sativum (pea), the PSI structure has been resolved up to 3.4 Å resolution as a photosystem I and light-harvesting complex (LHCI; collectively PSI–LHCI; Ben-Shem et al., 2003). Cyanobacterial PSI contains 12 subunits with 96 chlorophyll (Chl) cofactors, while the plant complex consists of at least 17 subunits harboring over 170 Chls. In cyanobacteria, PSI is mostly observed as a trimer of monomeric PSI cores (Kruip et al., 1994; Fromme et al., 2001), while PSI in plants, red, and green algae is monomeric (Scheller et al., 2001; Kouril et al., 2005). Two functional moieties that can be distinguished in PSI are the photosystem I core that includes the redox active cofactors, and the peripheral light-harvesting complex (LHCI), which serves to increase the absorption cross section (Schmid et al., 1997; Amunts et al., 2009). While the structural organization of the redox centers is virtually identical in the structures obtained from Pisum sativum and Synechococcus elongatus (Jordan et al., 2001; Amunts et al., 2007), the LHCI complex shows a high degree of variability in size, subunit composition, and number or type of bound pigments. This variation allows each organism to adjust to its specific natural habitat (Croce et al., 2007; Wientjes et al., 2009). The PSI core complex prepared from plants is sometimes also denoted as the PS1-110 particle, referring roughly to the total number (∼110) of bound Chls (Mullet et al., 1980) and has a molecular weight of ∼300 kDa.
As in PSII and bacterial reaction centers (RCs), the cofactors in PSI are symmetrically arranged in two parallel chains relative to a pseudo-C2 symmetry axis perpendicular to the membrane plane in which PSI is embedded in vivo (Fig. 1). Like type I bacterial RCs, PSI consists of six Chl cofactors, two quinones, and three iron–sulfur [4Fe−3S] clusters (FX, FA, FB) acting as intrinsic electron acceptors. The FA and FB clusters operate in series and are bound to the PsaC subunit. The FX cluster is located at the interface between the PsaA and PsaB subunits, while the accessory Chls (A−1A and A−1B), the Chl acceptors (A0A and A0B), and the quinone acceptors (A1A and A1B) are bound to the PsaA (A branch) and PsaB (B branch). In comparison to their PSII quinone counterparts, A1A and A1B in PSI are more tightly associated with the protein backbone and are not as readily accessible for chemical-reducing agents (Srinivasan and Golbeck, 2009). With distances ranging between 15 and 40 Å, the cofactors in the PSI RC are more isolated from the surrounding antenna pigments than the cofactors in PSII and bacterial RC.
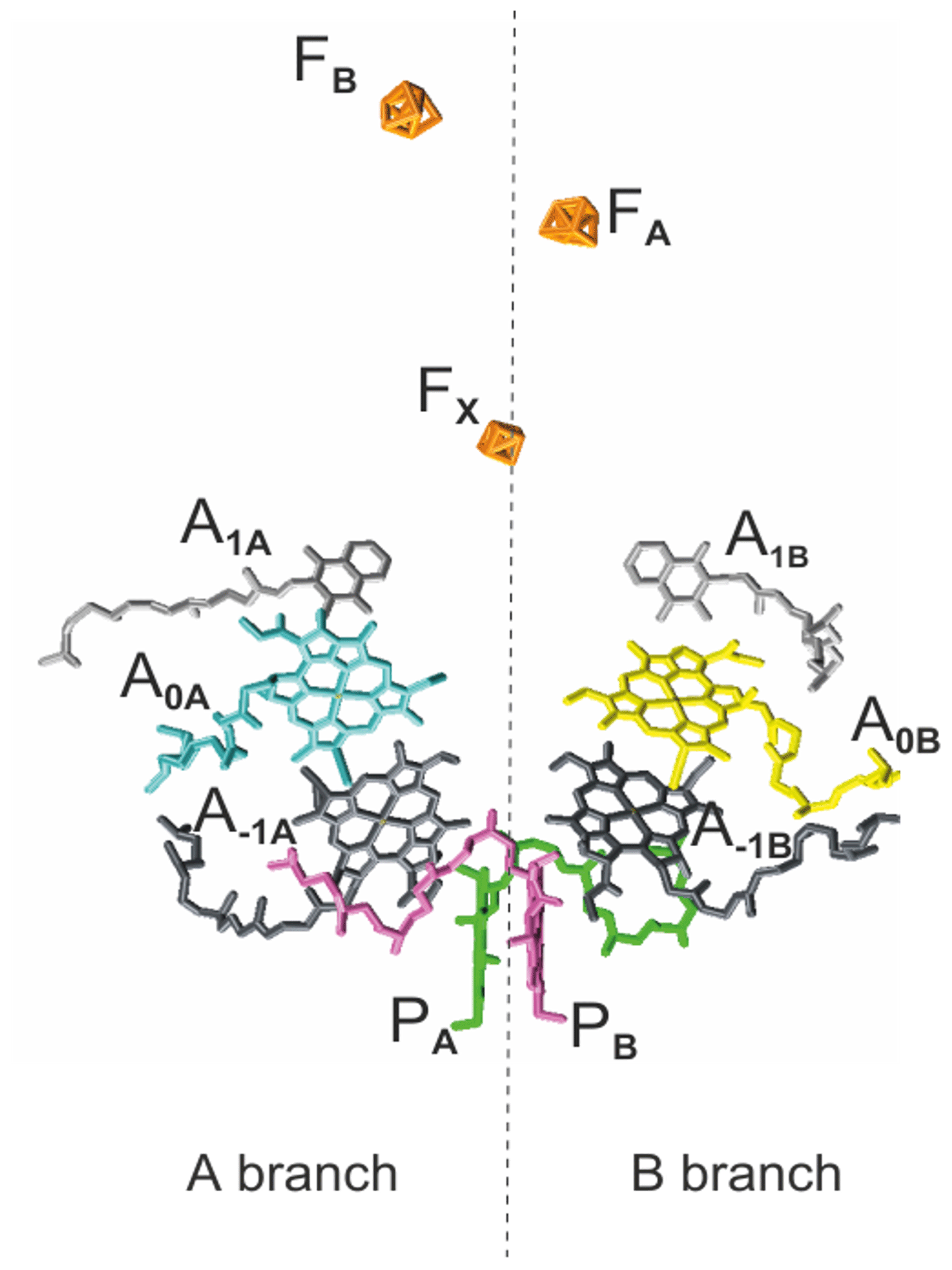
Figure 1The arrangement of cofactors in the PS1 RC. Depicted in pink and green are the two central Chls, PB and PA, of the Chl a ∕ Chl a' dimer. Furthermore, the RC contains two accessory Chl a (A−1A and A−1B), two donor Chl a (A0A and A0B), and two tightly associated phylloquinones (A1A and A1B). Finally, there are three iron–sulfur [4Fe−3S] clusters and FB) which function as terminal intrinsic electron acceptors. Both the A and B branches participate in electron transfer, with the relative activity depending mainly on the organism and the reduction conditions. PDB entry 2WSC (Amunts et al., 2009).
The electron donor, the heterodimeric P700, is, similarly to the FX cluster, located at the interface of both branches and consists of one chlorophyll a (Chl a; PB) and one Chl a' (PA), which is the C-132-epimer of Chl a. While PA forms hydrogen bonds to its protein environment, no hydrogen bonds are found on the PB side (Watanabe et al., 1985). The ratio of the spin-density distribution over the dimer exhibits significant diversity between species and conditions (Webber and Lubitz, 2001). Fourier transform infrared and electron paramagnetic resonance (EPR) spectroscopic studies on cyanobacterial PSI from Synechocystis indicated a ratio of electron spin density distribution in the range of 50:50 to 33:67 in favor of the PB (Breton et al., 1999). On the other hand, in spinach and Thermosynechococcus (T.) elongatus, ratios in the range of, respectively, 25:75–20:80 and 15:85 have been estimated (Davis et al., 1993; Käss et al., 2001). Electronic structure calculations suggested a ratio of 28:72 based on the coordinates taken from the high-resolution X-ray data of T. elongatus and indicate the hydrogen bonding of the PA Chl, the asymmetry in molecular geometry (Chl a / Chl a'), and minor differences in the protein environment as being the main factors influencing the relative spin density distribution over PA and PB (Saito et al., 2011).
Calculations making use of the frozen density embedding (FDE) technique on the primary electron donor of PSI in S. elongatus, including a large part of the protein environment, resulted in 76 % of the spin density being localized on PB (Artiukhin et al., 2020). Using FDE instead of the conventional Kohn–Sham density functional theory (DFT) can improve the description of the interaction of the electron donor with the protein matrix and the spin localization, as it avoids certain problems arising from the self-interaction error. The calculated spin populations were in good agreement with the references (Saito et al., 2011) and available experimental data obtained with 13C photochemically induced dynamic nuclear polarization (photo-CIDNP) magic angle spinning (MAS) nuclear magnetic resonance (NMR; Alia et al., 2004).
While the donor in PSII is the strongest oxidizing agent known in living nature, P700 is optimized to provide a strong reducing force, which is required for the formation of nicotinamide adenine dinucleotide phosphate (NADPH). With a potential of approximately −1.2 V, P700•+ is probably the strongest reducing entity found in living systems (Ishikita et al., 2006).
Whereas in type II bacterial RCs and PSII electron transfer (ET) proceeds along only one of the two pseudosymmetric branches, over the past few years evidence has been accumulated to show that both branches are active in ET in PSI, resulting in a two-sided ET often denoted as bidirectional ET (see Santabarbara et al., 2010). Since femtosecond optical studies indicated the accessory Chl A−1 to be the primary electron donor (Holzwarth et al., 2006; Müller et al., 2010), the structural asymmetry of the P700 Chl a / Chl a' dimer is no longer a convincing argument against the participation of both branches in ET. A possible reason for the occurrence of the two-sided ET in type I systems as PSI and RCs of heliobacteria (Thamarath et al., 2012b) but not in type II systems as PSII and purple bacterial RC might be that the quinones in type II RCs function as a two-electron gate, with a mobile quinone on the inactive branch being used as a terminal acceptor (Müh et al., 2012). In type I systems, on the other hand, the iron–sulfur clusters act as terminal acceptors, while the quinone serves as an intermediary in electron transfer, making two-sided ET feasible. While consensus on the two-sided nature of ET in both prokaryotic and eukaryotic PSI has been reached (Fairclough et al., 2003; Redding et al., 2007), the molecular details controlling the ET pathways are not yet fully elucidated (Berthold et al., 2012). The relative activity of the two branches is in favor of the A branch but seems to vary among different organisms ranging from ∼3 to 2 in green algae (Holzwarth et al., 2006; Li et al., 2006) and ∼3–4 to 1 in cyanobacteria (Ramesh et al., 2004; Dashdorj et al., 2005). The relation between the activity of the ET pathways and the electron (spin) density distribution between the two parts of the donor is not understood. In addition, the reducing conditions of the quinones appear to affect the relative branch activity with, e.g., ET in Synechococcus lividus occurring solely along the B branch at low temperatures (100 K) and strongly reducing conditions (Poluektov et al., 2005). Hence, the factors inducing the initial asymmetry are not yet understood.
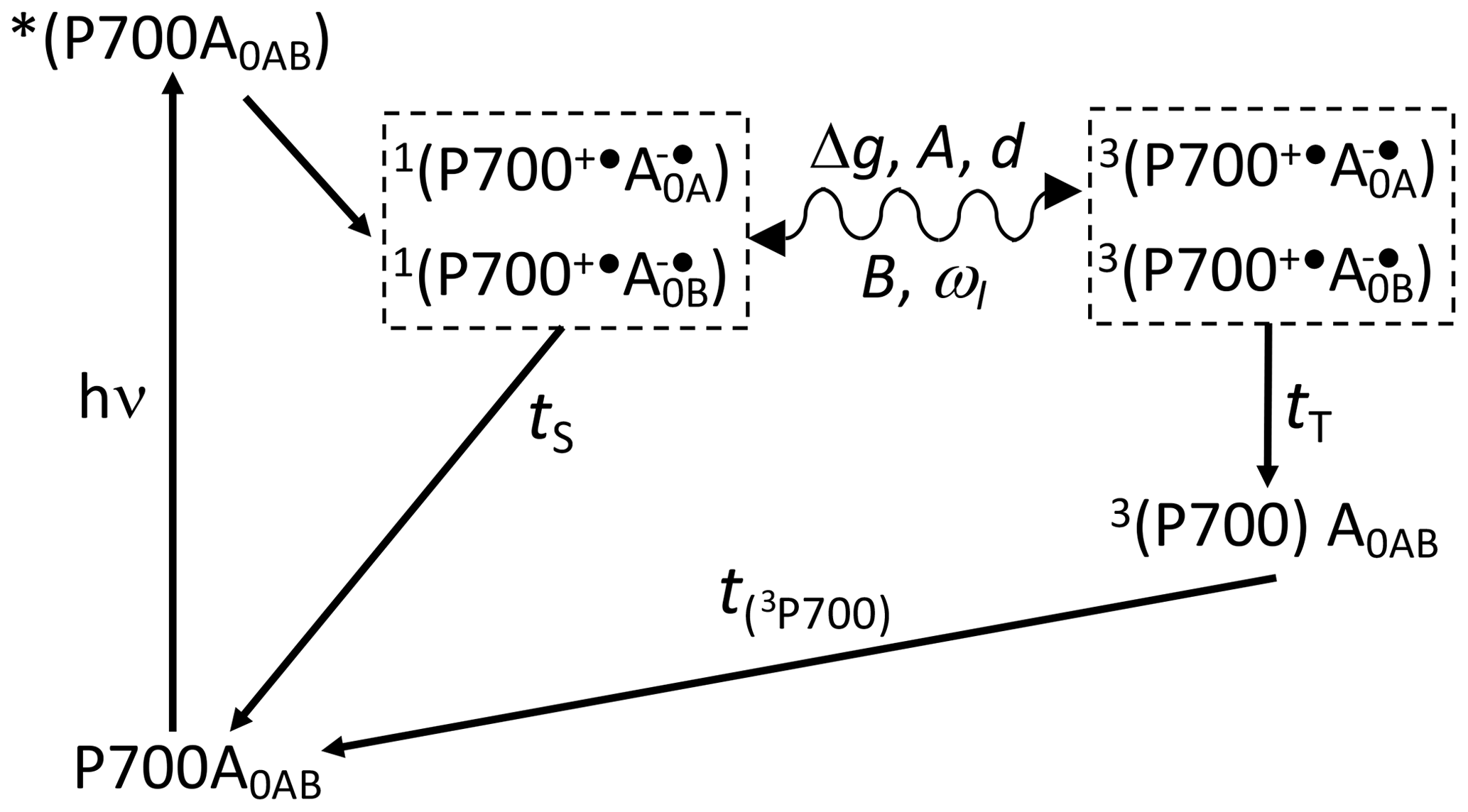
Scheme 1Reaction cycle in PSI, with reduced FX acceptor and electron transfer, over both branches of cofactors A and B. After the absorption of a photon, electron transfer occurs from the P700 donor to the primary acceptors A0. Upon chemical prereduction of FX, the electron transfer becomes cyclic. Due to spin conservation, the spin-correlated radical pair (SCRP) are formed in a pure singlet state. The SCRP in its singlet state can either recombine to the diamagnetic ground state or undergo coherent singlet–triplet interconversion to its electronic triplet state. This interconversion relies on the difference in the g values of the two electrons, Δg, and the hyperfine interaction with magnetic nuclei. According to the radical pair mechanism (RPM), this influence of the nuclei on the radical pair dynamics leads to spin sorting and, hence, to enrichment of nuclear spin states in the two decay channels. In frozen samples, three-spin mixing (TSM) produces nuclear hyperpolarization based on the secular part of the hyperfine interaction, A, the coupling between the two electrons, d, the pseudosecular hyperfine coupling, B, and the nuclear Zeeman frequency, ωI. Furthermore, as caused by the different kinetics of the two decay channels (TS vs. TT), the differential decay (DD) mechanism for nuclear spin hyperpolarization occurs. From the triplet state of the SCRP, a molecular donor triplet state is formed, which decays with a triplet lifetime, T(3P700), of ∼3 µs (Polm and Brettel, 1998), making the occurrence of the differential relaxation (DR) mechanism unlikely.
To further investigate the functional symmetry breaking in PSI, we have studied isotope-labeled PSI-110 samples from duckweed with photo-CIDNP MAS NMR spectroscopy. Photo-CIDNP MAS NMR spectroscopy is an analytical method (for a review, see Matysik et al., 2009; Bode et al., 2013) informing on the molecules involved in spin-correlated radical pairs in both their electronic ground state (using NMR chemical shift information) and radical pair state (by photo-CIDNP intensities). The method is based on the solid-state photo-CIDNP effect, discovered in bacterial RCs in 1994 (Zysmilich and McDermott, 1994), occurring in spin-correlated radical pairs (SCRPs) in an immobile matrix upon cyclic ET. The NMR signal is enhanced by up to a factor of 80 000 (Thamarath et al., 2012a). The effect requires a cyclic reaction process that is introduced by the prereduction of the acceptor site. Scheme 1 shows such a reaction cycle for PSI. In the electronically excited state of the donor (P700*), an electron is transferred to the primary acceptor (A0). The radical pair is formed in the singlet state and undergoes intersystem crossing to its triplet state. Magnetic coupling to nuclear spins alters the intersystem crossing rates for different nuclear spin states, leading to nuclear spin sorting on the singlet and triplet recombination pathways. Although both pathways return to the same product (i.e., the ground state P700-A0), nuclear polarization is generated. The spin chemical mechanism has been probed by field-dependent (Thamarath et al., 2012a; Gräsing et al., 2017), time-resolved (Daviso et al., 2009a, 2010; Sai Sanker Gupta et al., 2014), and preparation-dependent (Matysik et al., 2000a; Daviso et al., 2011) experiments. Nuclear polarization arises from several different mechanisms operating in parallel. The classical radical pair mechanism (RPM; Kaptein and Oosterhoff, 1969; Closs and Closs, 1969) relies on spin sorting and produces transient nuclear polarization in both branches, canceling on the arrival of the population of the slower triplet decay channel. In addition, electron–nuclear spin dynamics in the radical pair state induce nuclear spin polarization through two solid-state mechanisms called three-spin mixing (TSM; Jeschke, 1997) and differential decay (DD; Polenova and McDermott, 1999), which remains for the period given by the T1 relaxation time. Recently, Sosnovsky et al. (2016, 2019) reinterpreted these coherent solid-state mechanisms in terms of electron–electron–nuclear level crossings and level anticrossings. Furthermore, in the differential relaxation (DR) mechanism, also called cyclic reaction mechanism, the nuclear polarization of the triplet decay channel is quenched by the paramagnetic molecular triplet state, enhancing nuclear relaxation and making the cancellation of the RPM polarization incomplete (McDermott et al., 1998).
Various photosynthetic RCs of plants (Alia et al., 2004; Diller et al., 2005, 2007; Janssen et al., 2018), algae (Janssen et al., 2010, 2012), diatoms (Zill et al., 2017, 2019), purple bacteria (Prakash et al., 2007; Daviso et al., 2009b; Paul et al., 2019), heliobacteria (Thamarath et al., 2012b), green sulfur bacteria (Roy et al., 2008), and flavoproteins (Thamarath et al., 2010; Ding et al., 2019) have been analyzed with the photo-CIDNP MAS NMR method. Previously, the application of 13C photo-CIDNP MAS NMR was restricted to the unlabeled and isolated PSI complex due to difficulties in obtaining selective 13C labeling in plants. Based on the data obtained from natural abundance PSI, a first tentative assignment of the light-induced signals involved a single Chl a molecule, which is probably the PB cofactor of the donor P700 (Alia et al., 2004).
In this work, we report on the first selective incorporation of 13C isotope labels in a PSI complex from duckweed (Spirodela). Backed by 15N labeling and quantum chemical calculations, we have explored the photosynthetic machinery of PSI on 13C and 15N isotope-labeled preparations from duckweed by photo-CIDNP MAS NMR, aiming for the details of the electronic structure of the dimeric donor and the question of one- or two-sided ET. In addition to continuous illumination with white light, 13C photo-CIDNP MAS NMR was induced by a 532 nm nanosecond flash laser.
2.1 Photosystem I particle preparation
Duckweed plants were grown under aseptic conditions on half-strength Hunter's medium (Posner, 1967). For selective 13C labeling of plants, 1.4 mM of δ-aminolevulinic acid, isotopically 13C labeled at carbon position 4 (4-ALA; Cambridge Isotope Laboratories, Inc.), was added to the duckweed growth medium (half-strength Hunter's medium; pH 4.8). Plants were grown on labeled medium, under continuous light (20 ), at 25 ∘C. The medium was continuously bubbled with sterile air containing 5 % CO2. After 7 d, plants were harvested and used directly for sample preparation or frozen in liquid nitrogen and stored at −80 ∘C until use. The PSI complex containing ∼110 Chl / P700 (PS1-110 particles) was prepared according the method described by Alia et al. (2004).
2.2 Determination of the 15N and 13C label incorporation
Chl a were extracted from plants grown in ALA-supplemented half-strength Hunter's medium (labeled sample) and from unlabeled plants (reference sample), according to the procedure of Moran and Porath (1980). Plants were homogenized in MeOH. The methanolic solution was centrifuged for 5 min at 300×g. The green supernatant was separated and dried under a gentle stream of N2. The sample was resuspended in acetone, loaded on a cellulose column, and pure Chl a fractions were eluted with petroleum ether/acetone (7/3 v∕v). The solvent was evaporated under a N2 flow, and the pure Chl a was stored at −20 ∘C in a dry nitrogen atmosphere. Label incorporation has been determined by mass spectrometry to be about 75 % for each particular carbon position of the 4-ALA isotope label pattern. For details, see the Supplement.
2.3 Photo-CIDNP MAS NMR experiments
The NMR experiments were performed by using DMX/AV-100, DMX/AV-200, DMX/AV-300 and DMX/AV-400 NMR spectrometers (Bruker GmbH, Karlsruhe, Germany). The samples were loaded into optically transparent 4 mm sapphire rotors. The PSI samples were reduced by the addition of an aqueous solution of a 10 mM sodium dithionite solution prepared in a 40 mM glycine buffer (pH 9.5) in an oxygen-free atmosphere. Immediately following the reduction, slow freezing of the sample was performed directly in the NMR probe inside the magnet under continuous illumination with white light. All spectra have been obtained at a sample temperature of 235 K and with a spinning frequency of 8 kHz.
The spectra were collected with a spin echo pulse sequence with a phase cycle of (π∕2) pulses under two-pulse phase modulation (TPPM) carbon–proton decoupling (Bennett et al., 1995). Photo-CIDNP MAS NMR spectra have been obtained using continuous illumination with a 1000 W Xenon arc lamp (Matysik et al., 2000b). The number of scans was 20 000, unless stated differently. The fitting of the collected spectra was performed using Igor Pro 6.01 (WaveMetrics, Inc., Portland, OR, USA). Based on the relative intensity of the signals, the electron spin density was calculated for the nitrogen assigned to the donor.
A pulsed nanosecond flash laser provides sufficient radiation intensity for time-resolved photo-CIDNP MAS NMR studies and does not decrease the time resolution that can be obtained in NMR experiments. The laser is operating with a repetition rate between 1 and 10 Hz. Using 1064 nm flashes of a Nd:YAG laser (Spectra-Physics Quanta-Ray INDI 40-10; Irvine, CA, USA) upon frequency doubling with a second harmonic generator (SHG), 532 nm laser flashes with pulse length of 6–8 ns and an energy between 20 and 150 mJ are produced.
2.4 Quantum chemical calculations
The structural models employed in our calculations were extracted from the crystal structure of PSI in plants (PDB entry 2WSC; Amunts et al., 2010), provided by the Protein Data Bank (Berman et al., 2000). Two different types of molecular models were considered, namely the so-called ISO models corresponding to the isolated cofactors extracted from the crystal structure. The binding pocket models, abbreviated as r32 and r34, were created by specifying radii of 3.2 and 3.4 Å around each atom of the cofactor of interest. All surrounding cofactors, water molecules, and amino acid residues with at least one atom within these radii were included explicitly into these models. For geometry optimizations, the DFTB3 (density-functional tight-binding, extended version) (Gaus et al., 2012) method within the AMS-DFTB (Amsterdam Modeling Suite) module from the ADF (Amsterdam Density Functional) 2019 package (Amsterdam; Velde et al., 2001) was used. The third-order parametrization for organic and biological system (3ob; Gaus et al., 2013; Kubillus et al., 2015) parameters from the corresponding Slater–Koster file were used. The optimizations were performed as a sequence of several steps that partly optimize the protein structure. For further details on the model setup and geometry optimization, see Sect. S2.1 and S2.2 in the Supplement. Graphical examples of the generated structures can be found in Sect. S5 in the Supplement.
NMR calculations of the binding pocket models of r32 or r34 were carried out within a subsystem DFT approach (Jacob and Visscher, 2006), using the TZP (triple-zeta polarized) (van Lenthe and Baerends, 2003) basis set and the Perdew–Wang (referred to as PW91 hereafter; Perdew and Wang, 1991; Perdew et al., 1992) XC (exchange—correlation) functional with the conjoint (Lee et al., 1991) kinetic energy functional Perdew–Wang kinetic (PW91k; Lembarki and Chermette, 1994). 15N chemical shifts were calculated with respect to the ammonia shieldings, while 13C chemical shifts were calculated with respect to tetramethylsilane (TMS; for further details, see Sect. S2.3 in the Supplement). Ring current effects of other subsystems were considered by calculating nuclear independent chemical shifts (NICSs), following Jacob and Visscher (2006). For further details on the NMR calculations, see Sect. 2.3–2.5.
3.1 15N photo-CIDNP MAS NMR
Figure 2 shows 15N MAS NMR spectra of uniformly 15N labeled PSI-110 particles of duckweed obtained under continuous illumination with white light at magnetic field strengths of (a) 2.35, (b) 4.7, (c) 7.1, and (d) 9.4 Tesla. At higher fields, the signal of the amide backbone nitrogen of the protein becomes clearly visible at about 125 parts per million (ppm) as a broad peak. In addition, sharp light-induced emissive (negative) signals were observed to originate from the Chl a and Chl a' cofactors involved in formation of a SCRP. All light-induced signals are emissive at all magnetic fields investigated, and the absolute intensity increases with the magnetic field strength. Previous numerical simulations suggest that the matching conditions of the enhancement mechanisms are best met at 9.4 Tesla (i.e., 400 MHz 1H frequency), leading to maximum signal enhancement (Roy et al., 2007). The three emissive 15N signals appear at 254 (strong, with shoulder at 250), 210 (very strong, with weak shoulder at 207) and 188 ppm (medium). The signals are in good agreement with previous 15N photo-CIDNP MAS NMR data of PSI from duckweed and spinach obtained at 4.7 T (Janssen et al., 2012) and can be conveniently assigned to a Chl a cofactor, with signals at 247.0, 189.4, 206.5 and 186.6 ppm in the solution of NMR for N-IV, N-III, N-II and N-I, respectively (Boxer et al., 1974). The strongest signal belongs to a single N-II, and the second strongest originates from the N-IV nitrogen. It is not clear whether the third signal occurring at 188 ppm originates from either N-I or N-III. Since shoulders and asymmetries occur, it appears that the signals originate from multiple cofactors. Emissive signals can arise from either donor or acceptor cofactors, and therefore, the sign cannot indicate the site of origin of signals. Since a chemical shift assignment would allow one to recognize whether the signals originate from donor or acceptor, and, if from the donor, whether from Chl a or Chl a', we performed quantum chemical calculations to estimate the chemical shifts of the different cofactors.
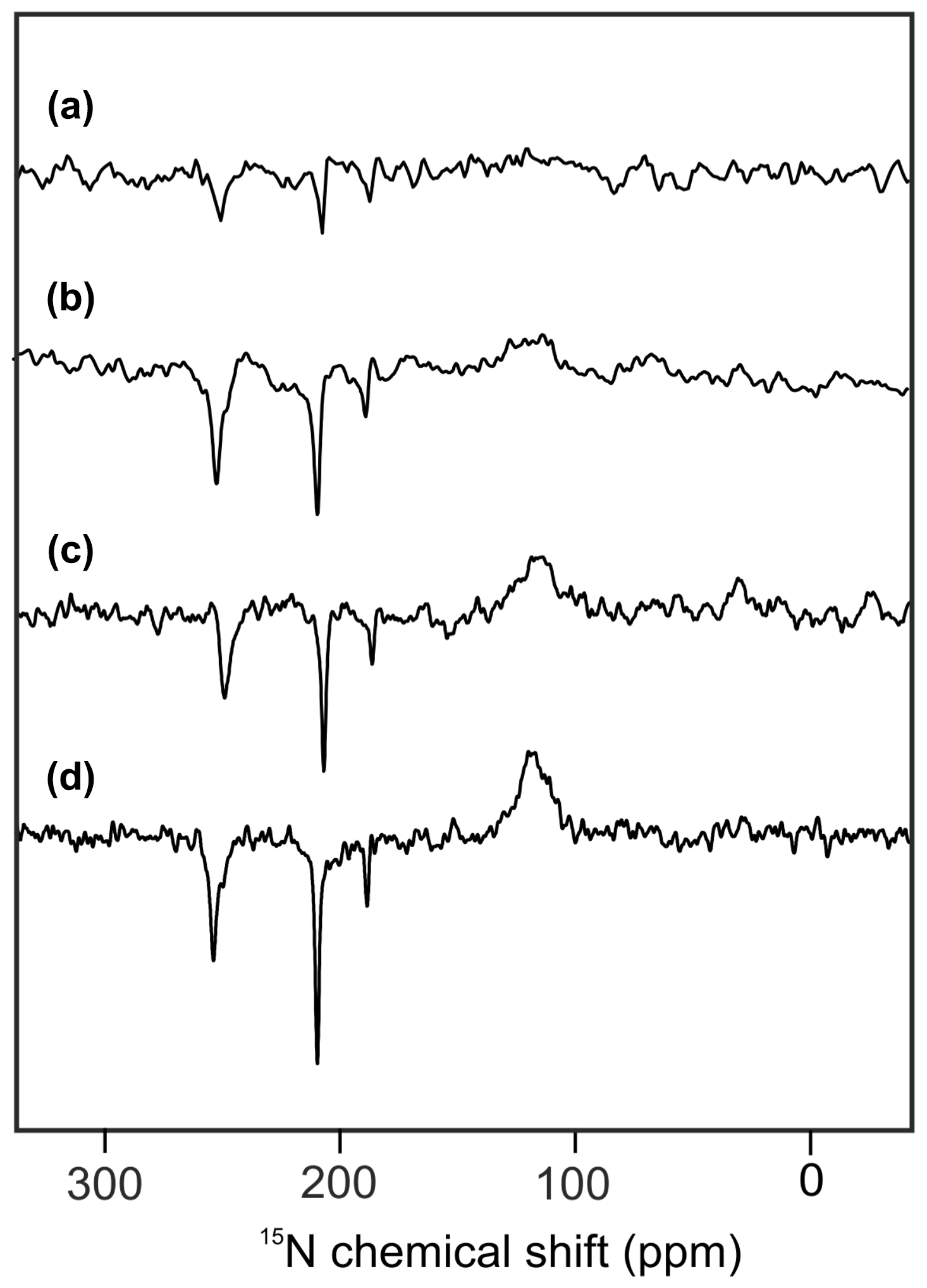
Figure 215N photo-CIDNP MAS NMR spectra obtained from the same sample of uniformly 15N labeled PSI-110 particles of duckweed measured at magnetic field strengths of (a) 2.35 T, (b) 4.7 T, (c) 7.1 T, and (d) 9.4 T. All spectra were obtained with a MAS frequency of 8 kHz, a temperature of 235 K, a cycle delay of 4 s, and an illuminance of 320 kLux provided by a white Xenon lamp. The number of scans was kept constant.
The calculated 15N chemical shifts of the two donor cofactors PA and PB, and the two acceptor cofactors A0A and A0B, are shown in Tables S2.1 and S2.2, respectively. The general chemical shift pattern is well reproduced by the calculations; however, assignment of resonances to specific cofactors is not possible. A possible source of the deviations from the calculated NMR shifts arises from the use of the static crystal structure rather than averaging over conformations accessible during the protein dynamics. This could be assessed by performing a short molecular dynamics (MDs) simulation and calculating NMR shifts for an ensemble of structures. Such a treatment could give an indication of thermal effects on the structure and, thus, implicitly on the NMR shifts. However, dynamic effects happening on longer timescales, which may be relevant for the NMR shifts as well, would not be covered in this way. The simulation of a structural ensemble is, however, beyond the scope of this work. Also, the inclusion of a sphere of protein environment of 3.2 and 3.4 Å (r32 and r34 in Table S2.1 and S2.2 and Fig. S3.1 to S3.4 in the Supplement) does not allow for a conclusive assignment. Although there is a significant environmental effect predicted, the strongest experimentally observed signal, N-II at 210 ppm, might be tentatively assigned either to PA or to A0B, with very similar values. The experimental signal at 254 ppm (N-IV) might be tentatively assigned to the donor cofactors with significantly larger chemical shifts than the acceptor signals. The two epimeric cofactors forming the donor cannot be distinguished on the basis of calculated chemical shifts. Hence, PSI, having four similar cofactors which might be involved into the formation of the SCRP, does not allow for straightforward 15N chemical shift assignment.
3.2 13C photo-CIDNP MAS NMR
To further characterize the individual cofactors of PSI involved in SCRP, we next performed 13C photo-CIDNP MAS NMR on selectively 13C labeled PSI. Previously, 13C photo-CIDNP MAS NMR studies on plant PSI have been restricted to experiments on unlabeled preparations due to the difficulty of incorporating selective 13C isotope labels into plant RCs. In the present study, we succeeded in selectively incorporating 4-ALA in PSI from duckweed with an isotope enrichment of 75 % for each particular carbon position of the 4-ALA isotope label pattern (Fig. 3A).
The 13C NMR spectra in Fig. 3B are obtained from 4-ALA-labeled PSI-110 preparations at a magnetic field strength of 4.7 T (1H frequency of 200 MHz; spectra a) and 9.4 T (400 MHz; spectra b) obtained under continuous light or in the dark. The spectra under illumination show several light-induced signals (shown in red) which are not observable in the dark (shown in black). The light-induced signature, however, is very different at the two magnetic fields. While the light-induced signals obtained at 4.7 T are entirely enhanced absorptive, at 9.4 T most of the signals appear emissive. Significant magnetic field effects have been observed for RCs of heliobacteria (Thamarath et al., 2012b) and purple bacteria (Thamarath et al., 2012a), and a similar dramatic sign change has been observed very recently in 13C MAS NMR spectra of natural abundance PSII preparations from the diatom Phaeodactylum tricornutum (Zill et al., 2019). For unlabeled PSI-110 preparations of spinach (Alia et al., 2004), an entirely emissive envelope has been observed at 400 MHz. In the present study, however, some signals appear to turn positive, suggesting that 13C isotope labeling indeed has some influence on the spin dynamics. The entirely emissive envelope observed at higher fields suggests the absence of contributions by the DR mechanism, which is reasonable due to the presence of carotenoids, implying that the solid-state photo-CIDNP effect relies on DD and TSM mechanisms. Since the ratio between TSM and DD is field dependent (Jeschke and Matysik, 2003), our data suggest that the TSM, expected to cause emissive signals as found for samples at natural abundance (Prakash et al., 2005), contributes more strongly, while the DD decays at higher fields.
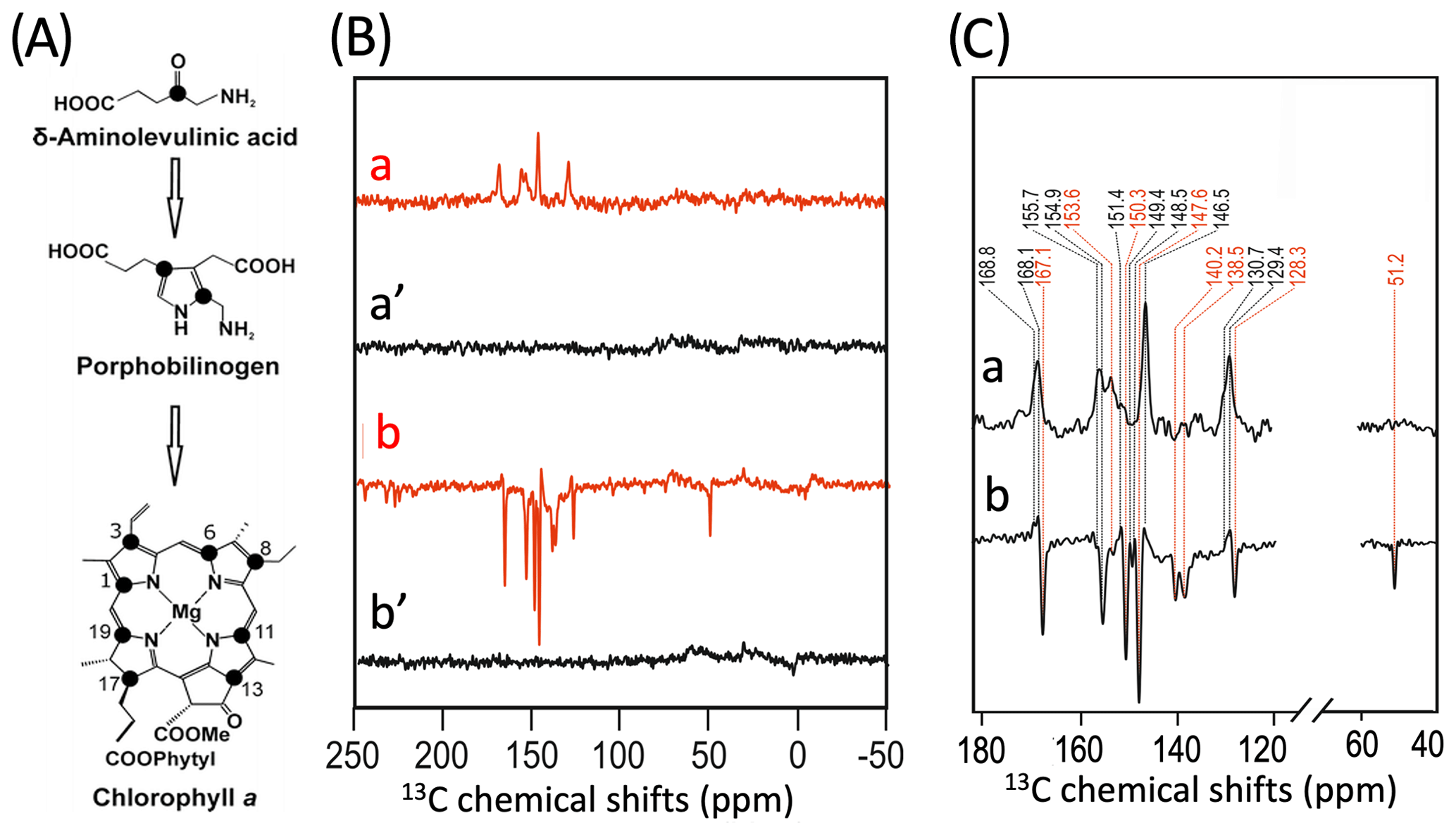
Figure 3(A) Incorporation of into cofactors (e.g., Chl a) of PS1-110 of duckweed. The black dots indicate the positions of 13C isotopes. (B) 13C photo-CIDNP MAS NMR spectra obtained under continuous illumination (red) of 4-ALA labeled PS1-110 particles of duckweed at a magnetic field strength of 4.7 T (a) and 9.4 T (b). Spectra (a', b') depicted in black originate from the corresponding experiments obtained in the dark. (C) Enlarged view of 13C photo-CIDNP MAS NMR spectra of 4-ALA-labeled PS1-110 particles of duckweed obtained at 4.7 T (a) and 9.4 T (b). Assigned signals are shown by the dashed lines, and emissive signals are in red.
A more detailed view of the light-induced signals is provided in Fig. 3C. The chemical shifts of the observed lines in the spectra are listed in Table 1. Careful examination of the spectra show that the emissive signals observed at a higher field (spectrum 3B in red) are canceled at a lower field, while the positive signals are visible in both spectra (in black). Therefore, it appears that the signals belong to two different sets. One might assume that one set originates from the donor and the other from the acceptor cofactors of PSI. Since the solid-state photo-CIDNP mechanisms DD and TSM require hyperfine anisotropy and occur on aromatic carbons, the occurrence of the emissive signal at 51.2 ppm at 9.4 T, originating from a C-17, the only aliphatic labeled position in the label pattern, is due to spin diffusion, i.e., polarization transfer from nearby 13C-labeled aromatic carbons. The cancellation of this signal at 4.7 T might imply that at that field the nearby aromatic carbons also do not obtain enhancement.
Table 113C chemical shifts of the photo-CIDNP signals obtained at 4.7 and 9.4 Tesla in comparison to literature data. Assignments obtained from 4-ALA-labeled samples are given in italics. See the footnotes below the table for more details. In the reference work, carbons C-9 and C-11 could not be separated.
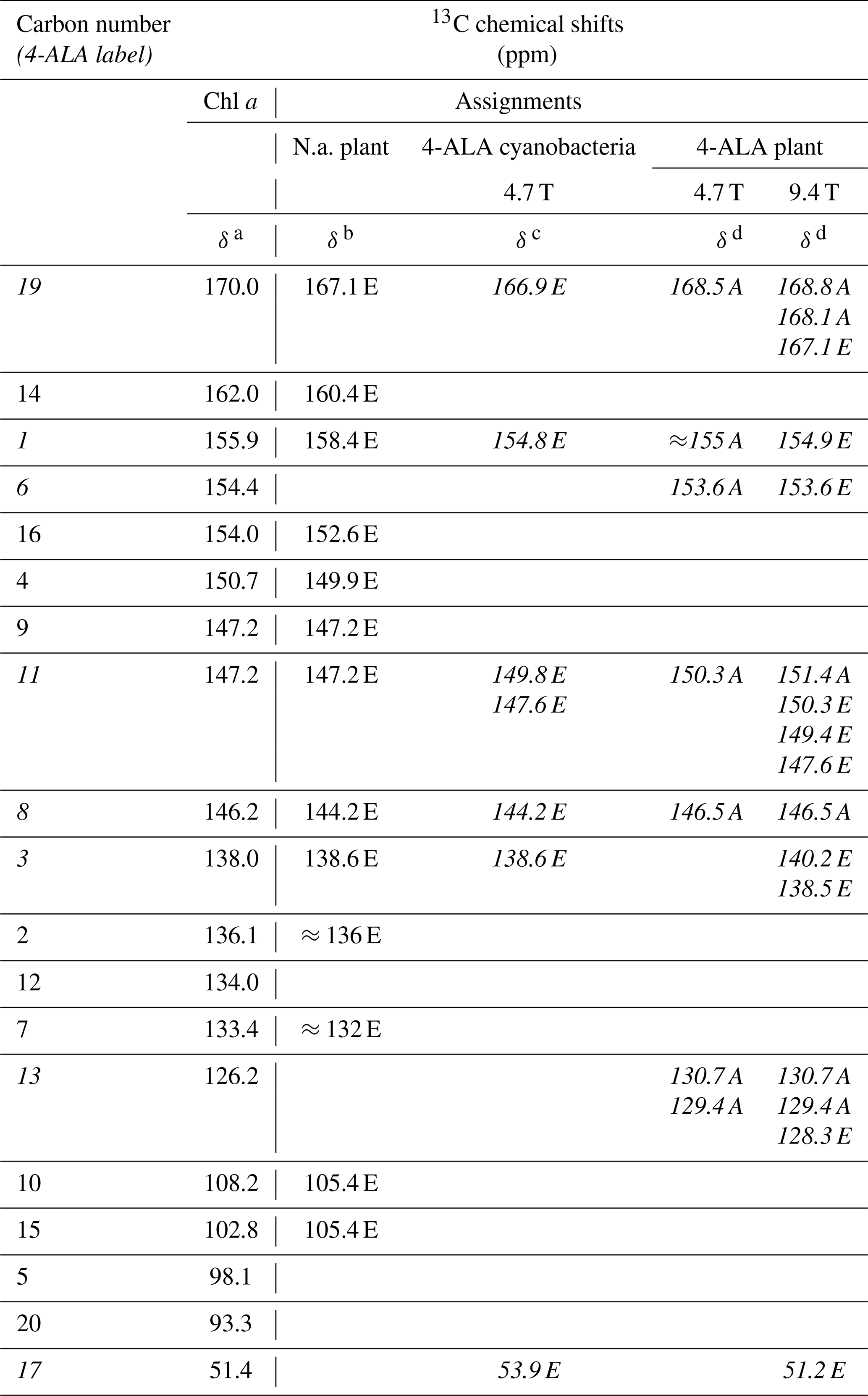
a Boender et al. (1995) – data experimentally obtained from solid aggregates of Chl a. b Alia et al. (2004) – data experimentally obtained from isolated PSI particles from spinach. c Janssen et al. (2010) – data experimentally obtained from 4-ALA labeled whole cells of Synechocystis containing both PSI and PSII. d This work. Data experimentally obtained from 4-ALA labeled isolated PSI from duckweed. A – absorptive (positive); E – emissive (negative) signal intensity; N.a. – natural abundant isotope distribution; ppm – parts per million; numbers in italics – 13C isotopically labeled in the 4-ALA pattern (Fig. 3).
All light-induced signals can be assigned conveniently to respective 13C-labeled carbon positions of cofactors (indicated in italics in Table 1). Since there is no light-induced signal which requires assignment to a nonlabeled position, we assume that all observed signals originate from 13C-labeled carbons. This assumption is reasonable considering the enrichment factor of 75 %. For several of the labeled 13C positions, multiple signals are observed which support the conclusion from the 15N data that several cofactors are observed. Three signals can be assigned to the carbons C-19 and C-13. For position C-11, four signals can be resolved. This observation strongly suggests that all four cofactors experience signal enhancement, implying that all four cofactors are involved in the spin-correlated radical pair and confirming that both electron transfer pathways are active. The alternating sign of the signals is typical for the magnetic field strength close to a turning point (see above).
To explore whether the assignment can be improved by attribution to individual cofactors occurring from the aromatic 13C carbons, quantum chemical calculations have been performed for the bare cofactors and have included surrounding amino acids up to a shell of 3.4 Å (Table S2.3 and S2.4). For C-11, four experimental values of 151.4, 150.3, 149.4, and 147.6 ppm have been observed. The calculated shifts for C-11 span a similar range of about 5 ppm. In general, the calculated values for the other carbon positions confirm this finding. The differences between the four cofactors are in the range of about less than 5 ppm in PSI. The differences in chemical shifts between the two BChl cofactors of the special pair in R. sphaeroides were found to be slightly larger in previous studies (Schulten et al., 2002; Daviso et al., 2009b; Sai Sankar Gupta et al., 2014). However, this relatively limited difference in chemical-shift asymmetry cannot explain the fundamentally different functional asymmetry in the bacterial RC. Since the chemical shift refers essentially to time-averaged electronic ground state properties, it is tempting to conclude that the different behavior of donor dimers is encoded in the dynamic structure. This is corroborated by studies of the functional symmetry breaking involving the special pair in bacterial RCs, which is thought to originate from specific long-living cooperative modes for the semiclassical coherent mixing of the charge transfer character into the electronically excited state from which the electron is transferred (Thamarath et al., 2012a) and local differences in molecular dynamics affecting the electron–phonon coupling (e.g., Novoderezhkin et al., 2004; Wawrzyniak et al., 2011).
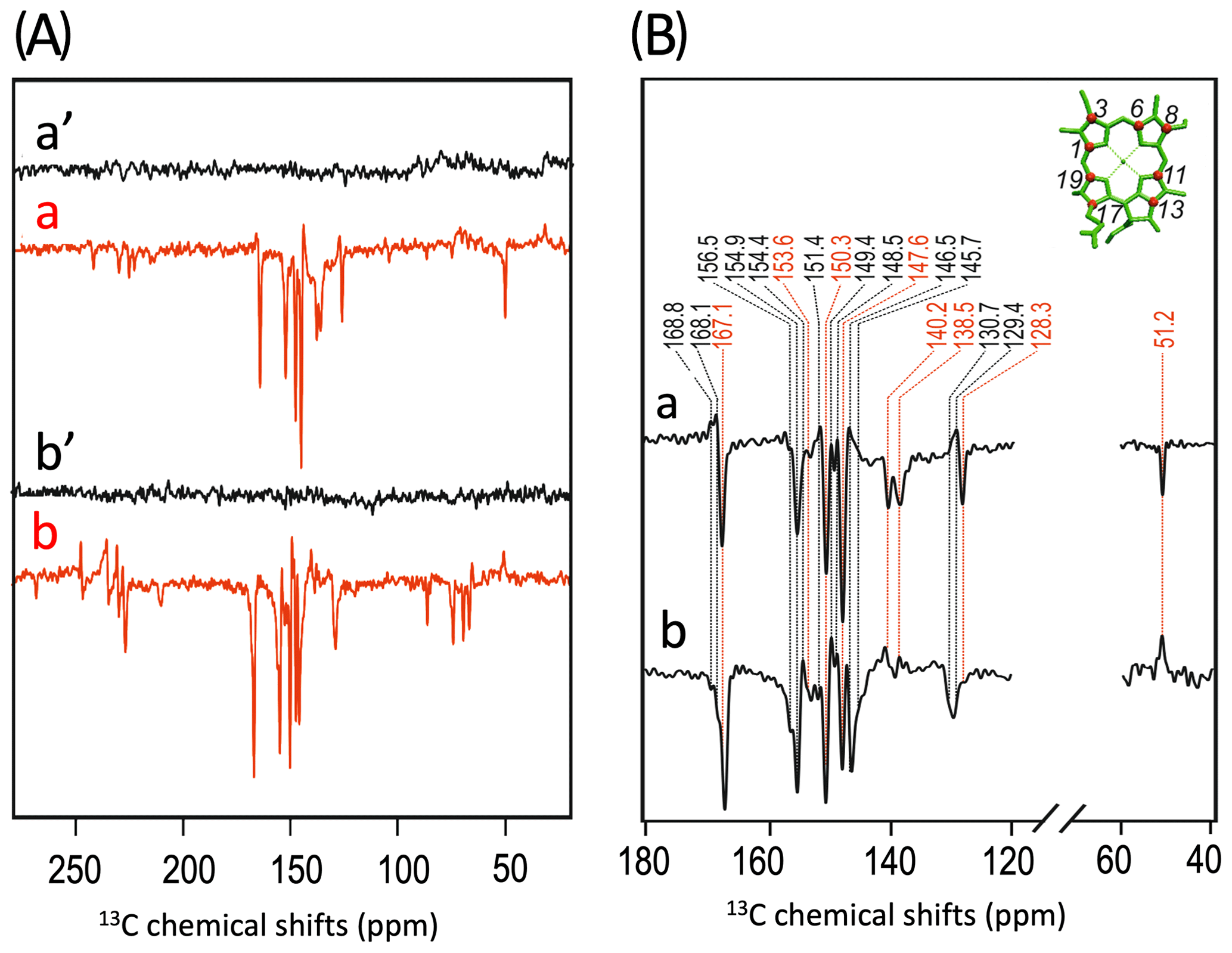
Figure 4(A) 13C photo-CIDNP MAS NMR spectra of 4-ALA-labeled PS1-110 particles of duckweed obtained at 9.4 T under continuous illumination (a) and under nanosecond laser flashes with zero delay (b). Spectra (a', b') depicted in black originate from the corresponding experiments obtained in the dark. (B) Enlarged region of 13C photo-CIDNP MAS NMR spectra of 4-ALA-labeled PS1-110 particles of duckweed obtained at 9.4 T under continuous illumination (a) and under nanosecond laser flashes with zero delay (b). Assigned signals are shown by the dashed lines, and emissive signals are in red.
Figure 4 compares the 13C photo-CIDNP MAS NMR spectrum induced by white light upon continuous illumination (spectrum 4Aa) with that induced by a 532 nm nanosecond flash laser (spectrum 4Ab). The magnified view of both light-induced spectra is shown in Fig. 4B. Remarkably, in the spectrum obtained by the laser flash experiment, several signals changed their sign. Enhanced absorptive signals turned emissive for the peaks at 168.8, 168.1 (both are assigned to C-19) and 129.4 ppm (C-13). On the other hand, several emissive signals become enhanced absorptive at 140.2, 138.5 (both arise from a C-3), and at 51.2 ppm (C-17). The different intensity patterns are due to differences in the enhancement mechanisms. Under steady-state illumination, the solid-state mechanisms TSM and DD produce the hyperpolarization which rely on anisotropic hyperfine interactions. In time-resolved experiments, the first detectable signals mainly refer to the singlet branch of the RPM and are based on isotropic hyperfine interactions. Equilibration of the polarization between the labeled carbons by spin diffusion occurs on a slower timescale (Daviso et al., 2009a).
There is experimental evidence that both the cofactors of the donor (PA and PB) and both the potential acceptor cofactors (A0A and A0B) carry the electron spin density of the spin-correlated radical pair. This confirms that both electron pathways in the PSI of duckweed are active and that the electron transfer does not occur exclusively in one branch. In addition, the time-averaged ground state electron density, as measured by the chemical shift, varies to a similar extent as in the functionally asymmetric special pair of RCs of R. sphaeroides. Our study suggests that the breaking of functional symmetry is not primarily due to local variation in time-averaged electronic ground-state properties at the donor site but, for instance, local and global electronic excited-state properties in conjunction with molecular dynamics.
| ALA | δ-aminolevulinic acid |
| Chl | Chlorophyll |
| DD | Differential decay |
| DFT | Density functional theory |
| DR | Differential relaxation |
| ET | Electron transfer |
| LHCI | Light-harvesting complex |
| MAS | Magic angle spinning |
| NMR | Nuclear magnetic resonance |
| P | Primary donor |
| photo-CIDNP | Photochemically induced dynamic nuclear polarization |
| PS | Photosystem |
| RC | Reaction center |
| RPM | Radical pair mechanism |
| SCRP | Spin-correlated radical pair |
| TSM | Three-spin mixing |
Original NMR data were obtained before 2013 and cannot be provided.
The Supplement contains the following information: determination of the isotope incorporation, computational details, chemical shifts calculated by quantum chemical methods, the effect of the protein environment on the calculated NMR Shifts, and graphical examples of structures. The supplement related to this article is available online at: https://doi.org/10.5194/mr-1-261-2020-supplement.
HJMdG, JM, and AA designed the research, and GJJ and AA prepared the samples. GJJ measured the NMR spectra, PE, BEB, and JN provided the quantum chemical calculations, and GJJ, PK, BEB, JM, and AA interpreted the data. The paper was written with contributions from all the authors. All authors approved the final version of the paper.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Robert Kaptein Festschrift”. It is not associated with a conference.
The authors would like to thank Karthick Babu Sai Sankar Gupta, Fons Lefeber, and Kees Erkelens for their kind help and Peter Gast and Hans van Gorkom (Leiden University) for the stimulating discussions.
This research has been supported by the Dutch Science Organization (NWO (grant no. 818.02.019)), the Deutsche Forschungsgemeinschaft (grant nos. MA 497/2-1 and MA 497/11-1), the Alexander von Humboldt-Stiftung, and the People Programme (Marie Curie Actions) of the European Union, Seventh Framework Programme (grant agreement no. 252213).
This paper was edited by Rolf Boelens and reviewed by Peter Hore and Gunnar Jeschke.
Amsterdam density functional program: https://www.scm.com/, last access: 1 September 2020.
Alia, Roy, E., Gast, P., van Gorkom, H. J., de Groot, H. J. M., Jeschke, G., and Matysik, J.: Photochemically induced dynamic nuclear polarization in photosystem I of plants observed by 13C magic-angle spinning NMR, J. Am. Chem. Soc., 126, 12819–12826, https://doi.org/10.1021/ja048051+, 2004.
Amunts, A. and Nelson, N.: Plant photosystem I design in the light of evolution, Structure, 17, 637–650, https://doi.org/10.1016/j.str.2009.03.006, 2009.
Amunts, A., Drory, O., and Nelson, N.: The structure of a plant photosystem I supercomplex at 3.4 Å resolution, Nature, 447, 58–63, https://doi.org/10.1038/nature05687, 2007.
Amunts, A., Toporik, H., Borovikova, A., and Nelson, N.: Structure determination and improved model of plant photosystem I, J. Biol. Chem, 285, 3478–3486, https://doi.org/10.1074/jbc.M109.072645, 2010.
Artiukhin, D. G., Eschenbach, P., and Neugebauer, J.: Computational investigation of the spin-density asymmetry in photosynthetic reaction center models from first principles, J. Phys. Chem. B, 124, 4873–4888, https://doi.org/10.1021/acs.jpcb.0c02827, 2020.
Bennett, A. E., Rienstra, C. M., Auger, M., Lakshmi, K. V., and Griffin, R. G.: Heteronuclear decoupling in rotating solids, J. Chem. Phys., 103, 6951–6958, https://doi.org/10.1063/1.470372, 1995.
Ben-Shem, A., Frolow, F., and Nelson, N.: Crystal structure of plant photosystem I, Nature, 426, 630–635, https://doi.org/10.1038/nature02200, 2003.
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., and Bourne, P. E.: The protein data bank, Nucleic Acids Res., 28, 235–242, https://doi.org/10.1093/nar/28.1.235, 2000.
Berthold, T., von Gromoff, E. D., Santabarbara, S., Stehle, P., Link, G., Poluektov, O. G., Heathcote, P., Beck, C. F., Thurnauer, M. C., and Kothe, G.: Exploring the Electron Transfer Pathways in Photosystem I by High-Time-Resolution Electron Paramagnetic Resonance: Observation of the B-Side Radical Pair PA in Whole Cells of the Deuterated Green Alga Chlamydomonas reinhardtii at Cryogenic Temperatures, J. Am. Chem. Soc., 134, 5563–5576, https://doi.org/10.1021/ja208806g, 2012.
Bode, B. E., Thamarath, S. S., Sai Sankar Gupta, K. B., Alia, A., Jeschke, G., and Matysik, J.: The solid-state photo-CIDNP effect and its analytical application, in: Hyperpolarization methods in NMR spectroscopy, edited by: Kuhn, L., Springer Berlin-Heidelberg, 105–121, 2013.
Boender, G. J., Raap, J., Prytulla, S., Oschkinat, H., and de Groot, H.: MAS NMR structure refinement of uniformly 13C enriched chlorophyll a/water aggregates with 2D dipolar correlation spectroscopy, Chem. Phys. Lett., 237, 502–508, https://doi.org/10.1016/0009-2614(95)00357-A, 1995.
Boxer, S. G., Closs, G. L., and Katz, J. J.: Effect of magnesium coordination on the carbon-13 and nitrogen-15 magnetic resonance spectra of chlorophyll a. Relative energies of nitrogen n-pi* states as deduced from a complete assignment of chemical shifts, J. Am. Chem. Soc., 96, 7058–7066, https://doi.org/10.1021/ja00829a038, 1974.
Breton, J., Nabedryk, E., and Leibl, W.: FT-IR study of the primary electron donor of photosystem I (P700) revealing delocalization of the charge in P700+ and localization of the triplet character in 3P700, Biochemistry-US, 38, 11585–11592, https://doi.org/10.1021/bi991216k, 1999.
Closs, G. L. and Closs, L. E.: Induced dynamic nuclear spin polarization in reactions of photochemically and thermally generated triplet diphenylmethylene, J. Am. Chem. Soc., 91, 4549–4550, https://doi.org/10.1021/ja01044a041, 1969.
Croce, R., Chojnicka, A., Morosinotto, T., Ihalainen, J. A., van Mourik, F., Dekker, J. P., Bassi, R., and van Grondelle, R.: The low-energy forms of photosystem I light-harvesting complexes: spectroscopic properties and pigment-pigment interaction characteristics, Biophys. J., 93, 2418–2428, https://doi.org/10.1529/biophysj.107.106955, 2007.
Dashdorj, N., Zhang, H., Kim, H., Yan, J., Cramer, W. A., and Savikhin, S.: The single chlorophyll a molecule in the cytochrome b6f complex: unusual optical properties protect the complex against singlet oxygen, Biophys. J., 88, 4178–4187, https://doi.org/10.1529/biophysj.104.058693, 2005.
Davis, I. H., Heathcote, P., MacLachlan, D. J., and Evans, M. C.: Modulation analysis of the electron spin echo signals of in vivo oxidised primary donor 14N chlorophyll centres in bacterial, P870 and P960, and plant Photosystem I, P700, reaction centres, BBA-Bioenergetics, 1143, 183–189, https://doi.org/10.1016/0005-2728(93)90141-2, 1993.
Daviso, E., Alia, A., Prakash, S., Diller, A., Gast, P., Lugtenburg, J., Matysik, J., and Jeschke, G.: Electron−nuclear spin dynamics in a bacterial photosynthetic reaction center, J. Phys. Chem. C, 113, 10269–10278, https://doi.org/10.1021/jp900286q, 2009a.
Daviso, E., Prakash, S., Alia, A., Gast, P., Neugebauer, J., Jeschke, G., and Matysik, J.: The electronic structure of the primary electron donor of reaction centers of purple bacteria at atomic resolution as observed by photo-CIDNP 13C NMR, P. Natl. Acad. Sci. USA, 106, 22281–22286, https://doi.org/10.1073/pnas.0908608106, 2009b.
Daviso, E., Prakash, S., Alia, A., Gast, P., Jeschke, G., and Matysik, J.: Nanosecond-flash 15N Photo-CIDNP MAS NMR on reaction centers of Rhodobacter sphaeroides R26, Appl. Magn. Reson., 37, 49–63, https://doi.org/10.1007/s00723-009-0050-2, 2010.
Daviso, E., Janssen, G. J., Alia, A., Jeschke, G., Matysik, J., and Tessari, M.: A 10,000-fold nuclear hyperpolarization of a membrane protein in the liquid phase via a solid-state mechanism, J. Am. Chem. Soc., 133, 16754–16757, https://doi.org/10.1021/ja206689t, 2011.
Diller, A., Alia, Gast, P., van Gorkom, H. J., Zaanen, J., de Groot, H. J. M., Glaubitz, C., and Matysik, J.: The electronic structure of the primary donor of photosystem II investigated by photo-CIDNP ssNMR, in: Photosynthesis: Fundamental Aspects to Global Perspectives, Proceedings of the 13th International Congress on Photosynthesis, Montreal, Canada, 29 August to 3 September 2004, edited by: van der Est, A. and Bruce, D., Allen Press, 307–308, 2005.
Diller, A., Roy, E., Gast, P., van Gorkom, H. J., de Groot, H. J. M., Glaubitz, C., Jeschke, G., Matysik, J., and Alia, A.: 15N photochemically induced dynamic nuclear polarization magic-angle spinning NMR analysis of the electron donor of photosystem II, Proc. Natl. Acad. Sci. USA, 104, 12767–12771, https://doi.org/10.1073/pnas.0701763104, 2007.
Ding, Y., Kiryutin, A. S., Yurkovskaya, A. V., Sosnovsky, D. V., Sagdeev, R. Z., Bannister, S., Kottke, T., Kar, R. K., Schapiro, I., Ivanov, K. L., and Matysik, J.: Nuclear spin-hyperpolarization generated in a flavoprotein under illumination: experimental field-dependence and theoretical level crossing analysis, Sci. Rep.-UK, 9, 18436, https://doi.org/10.1038/s41598-019-54671-4, 2019.
Emerson, R. and Chalmers, R. V.: Speculations concerning the function and phylogenetic significance of the assessory pigments in algae, Phycol. Soc. Amer. News Bull., 11, 51–56, 1958.
Fairclough, W. V., Forsyth, A., Evans, M. C., Rigby, S. E., Purton, S., and Heathcote, P.: Bidirectional electron transfer in photosystem I: Electron transfer on the PsaA side is not essential for phototrophic growth in Chlamydomonas, BBA-Bioenergetics, 1606, 43–55, https://doi.org/10.1016/S0005-2728(03)00083-5, 2003.
Fromme, P., Jordan, P., and Krauß, N.: Structure of photosystem I, BBA-Bioenergetics, 1507, 5–31, https://doi.org/10.1016/S0005-2728(01)00195-5, 2001.
Gaus, M., Cui, Q., and Elstner, M.: DFTB3: Extension of the self-consistent-charge density-functional tight-binding method (SCC-DFTB), J. Chem. Theory Comput., 7, 931–948, https://doi.org/10.1021/ct100684s, 2012.
Gaus, M., Goez, A., and Elstner, M.: Parametrization and benchmark of DFTB3 for organic molecules, J. Chem. Theory Comput., 9, 338–354, https://doi.org/10.1021/ct300849w, 2013.
Govindjee and Rabinowitch, E.: Two forms of chlorophyll a in vivo with distinct photochemical functions, Science, 132, 355–356, https://doi.org/10.1126/science.132.3423.355, 1960.
Gräsing, D., Bielytskyi, P., Céspedes-Camacho, I. F., Alia, A., Marquardsen, T., Engelke, F., and Matysik, J.: Field-cycling NMR with high-resolution detection under magic-angle spinning: determination of field-window for nuclear hyperpolarization in a photosynthetic reaction center, Sci. Rep.-UK, 7, 12111, https://doi.org/10.1038/s41598-017-10413-y, 2017.
Holzwarth, A. R., Müller, M. G., Niklas, J., and Lubitz, W.: Ultrafast transient absorption studies on photosystem I reaction centers from Chlamydomonas reinhardtii. 2: mutations near the P700 reaction center chlorophylls provide new insight into the nature of the primary electron donor, Biophys. J., 90, 552–565, https://doi.org/10.1529/biophysj.105.059824, 2006.
Ishikita, H., Biesiadka, J., Loll, B., Saenger, W., and Knapp, E.-W.: Cationic state of accessory chlorophyll and electron transfer through pheophytin to plastoquinone in photosystem II, Angew. Chem. Int. Ed., 118, 1998–1999, https://doi.org/10.1002/ange.200503804, 2006.
Jacob, C. R. and Visscher, L.: Calculation of nuclear magnetic resonance shieldings using frozen-density embedding, J. Chem. Phys., 125, 194104, https://doi.org/10.1063/1.2370947, 2006.
Janssen, G. J., Daviso, E., van Son, M., de Groot, H. J. M., Alia, A., and Matysik, J.: Observation of the solid-state photo-CIDNP effect in entire cells of cyanobacteria Synechocystis, Photosynth. Res., 104, 275–282, https://doi.org/10.1007/s11120-009-9508-1, 2010.
Janssen, G. J., Roy, E., Matysik, J., and Alia, A.: 15N Photo-CIDNP MAS NMR to reveal functional heterogeneity in electron donor of different plant organisms, Appl. Magn. Reson., 42, 57–67, https://doi.org/10.1007/s00723-011-0283-8, 2012.
Janssen, G. J., Bielytskyi, P., Artiukhin, D. G., Neugebauer, J., de Groot, H. J. M., Matysik, J., and Alia, A.: Photochemically induced dynamic nuclear polarization NMR on photosystem II: donor cofactor observed in entire plant, Sci. Rep.-UK, 8, 17853, https://doi.org/10.1038/s41598-018-36074-z, 2018.
Jeschke, G.: Electron–electron–nuclear three-spin mixing in spin-correlated radical pairs, J. Chem. Phys., 106, 10072–10086, https://doi.org/10.1063/1.474063, 1997.
Jeschke, G. and Matysik, J.: A reassessment of the origin of photochemically induced dynamic nuclear polarization effects in solids, Chem. Phys., 294, 239–255, https://doi.org/10.1016/S0301-0104(03)00278-7, 2003.
Jordan, P., Fromme, P., Witt, H. T., Klukas, O., Saenger, W., and Krauss, N.: Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution, Nature, 411, 909–917, https://doi.org/10.1038/35082000, 2001.
Kaptein, R. and Oosterhoff, J. L.: Chemically induced dynamic nuclear polarization II, Chem. Phys. Lett., 4, 195–197, https://doi.org/10.1016/0009-2614(69)80098-9, 1969.
Käss, H., Fromme, P., Witt, H. T., and Lubitz, W.: Orientation and electronic structure of the primary donor radical cation in photosystem I: A single crystals EPR and ENDOR study, J. Phys. Chem. B, 105, 1225–1239, https://doi.org/10.1021/jp0032311, 2001.
Kouril, R., van Oosterwijk, N., Yakushevska, A. E., and Boekema, E. J.: Photosystem I: a search for green plant trimers, Photochem. Photobiol. Sci., 4, 1091–1094, https://doi.org/10.1039/B505519A, 2005.
Kruip, J., Bald, D., Boekema, E., and Rögner, M.: Evidence for the existence of trimeric and monomeric Photosystem I complexes in thylakoid membranes from cyanobacteria, Photosynth. Res., 40, 279–286, https://doi.org/10.1007/BF00034777, 1994.
Kubillus, M., Kubař, T., Gaus, M., Řezáč, J., and Elstner, M.: Parameterization of the DFTB3 method for Br, Ca, Cl, F, I, K, and Na in organic and biological systems, J. Chem. Theory Comput., 11, 332–342, https://doi.org/10.1021/ct5009137, 2015.
Lee, H., Lee, C., and Parr, R. G.: Conjoint gradient correction to the Hartree-Fock kinetic- and exchange-energy density functionals, Phys. Rev. A, 44, 768–771, https://doi.org/10.1103/PhysRevA.44.768, 1991.
Lembarki, A. and Chermette, C.: Obtaining a gradient-corrected kinetic-energy functional from the Perdew-Wang exchange functional, Phys. Rev. A, 50, 5328–5331, https://doi.org/10.1103/PhysRevA.50.5328, 1994.
Li, Y., van der Est, A., Lucas, M. G., Ramesh, V. M., Gu, F., Petrenko, A., Lin, S., Webber, A. N., Rappaport, F., and Redding, K.: Directing electron transfer within Photosystem I by breaking H-bonds in the cofactor branches, P. Natl. Acad. Sci. USA, 103, 2144–2149, https://doi.org/10.1073/pnas.0506537103, 2006.
Matysik, J., Alia, Gast, P., van Gorkom, H. J., Hoff, A. J., and de Groot, H. J.: Photochemically induced nuclear spin polarization in reaction centers of photosystem II observed by 13C-solid-state NMR reveals a strongly asymmetric electronic structure of the P680(.+) primary donor chlorophyll, P. Natl. Acad. Sci. USA, 97, 9865–9870, https://doi.org/10.1073/pnas.170138797, 2000a.
Matysik, J., Alia, Hollander, J. G., Egorova-Zachernyuk, T., Gast, P., and de Groot, H. J. M.: Sample illumination and photo-CIDNP in a magic-angle spinning NMR probe, Indian J. Biochem. Bio., 37, 418–423, 2000b.
Matysik, J., Diller, A., Roy, E., and Alia: The solid-state photo-CIDNP effect, Photosynth. Res., 102, 427–435, https://doi.org/10.1007/s11120-009-9403-9, 2009.
McDermott, A., Zysmilich, M. G., and Polenova, T.: Solid state NMR studies of photoinduced polarization in photosynthetic reaction centers: Mechanism and simulations, Solid State Nucl. Mag., 11, 21–47, https://doi.org/10.1016/s0926-2040(97)00094-5, 1998.
Moran, R. and Porath, D.: Chlorophyll determination in intact tissues using n,n-dimethylformamide, Plant Physiol., 65, 478–479, https://doi.org/10.1104/pp.65.3.478, 1980.
Müh, F., Glöckner, C., Hellmich, J., and Zouni, A.: Light-induced quinone reduction in photosystem II, Biochim. Biophys. Ac., 1817, 44–65, https://doi.org/10.1016/j.bbabio.2011.05.021, 2012.
Müller, M. G., Slavov, C., Luthra, R., Redding, K. E., and Holzwarth, A. R.: Independent initiation of primary electron transfer in the two branches of the photosystem I reaction center, P. Natl. Acad. Sci. USA, 107, 4123–4128, https://doi.org/10.1073/pnas.0905407107, 2010.
Mullet, J. E., Burke, J. J., and Arntzen, C. J.: Chlorophyll proteins of photosystem I, Plant Physiol., 65, 814–822, https://doi.org/10.1104/pp.65.5.814, 1980.
Novoderezhkin, V. I., Yakovlev, A. G., van Grondelle, R., and Shuvalov, V. A.: Coherent nuclear and electronic dynamics in primary charge separation in photosynthetic reaction centers: A Redfield Theory approach, J. Phys. Chem. B, 108, 7445–7457, https://doi.org/10.1021/jp0373346, 2004.
Paul, S., Roy, U., Böckers, M., Neugebauer, J., Alia, A., and Matysik, J.: 15N photo-CIDNP MAS NMR analysis of a bacterial photosynthetic reaction center of Rhodobacter sphaeroides wildtype, J. Chem. Phys., 151, 195101, https://doi.org/10.1063/1.5128783, 2019.
Perdew, J. P. and Wang, Y.: Electronic structure of solids'91, Akademie, Berlin, 1991.
Perdew, J. P., Chevary, J. A., Vosko, S. H., Jackson, K. A., Pederson, M. R., Singh, D. J., and Fiolhais, C.: Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B, 46, 6671–6687, https://doi.org/10.1103/PhysRevB.46.6671, 1992.
Polenova, T. and McDermott, A. E.: A coherent mixing mechanism explains the photoinduced nuclear polarization in photosynthetic reaction centers, J. Phys. Chem. B, 103, 535–548, https://doi.org/10.1021/jp9822642, 1999.
Polm, M. and Brettel, K.: Secondary pair charge recombination in photosystem I under strongly reducing conditions: Temperature dependence and suggested mechanism, Biophys. J., 74, 3173–3181, https://doi.org/10.1016/S0006-3495(98)78023-3, 1998.
Poluektov, O. G., Paschenko, S. V., Utschig, L. M., Lakshmi, K. V., and Thurnauer, M. C.: Bidirectional electron transfer in photosystem I: direct evidence from high-frequency time-resolved EPR spectroscopy, J. Am. Chem. Soc., 127, 11910–11911, https://doi.org/10.1021/ja053315t, 2005.
Prakash, S., Alia, Gast, P., de Groot, H. J. M., Jeschke, G., and Matysik, J.: Magnetic field dependence of photo-CIDNP MAS NMR on photosynthetic reaction centers of Rhodobacter sphaeroides WT, J. Am. Chem. Soc., 127, 14290–14298, https://doi.org/10.1021/ja054015e, 2005.
Prakash, S., Alia, Gast, P., de Groot, H. J. M., Jeschke, G., and Matysik, J.: 13C chemical shift map of the active cofactors in photosynthetic reaction centers of Rhodobacter sphaeroides revealed by photo-CIDNP MAS NMR, Biochemistry, 46, 8953–8960, 2007.
Ramesh, V. M., Gibasiewicz, K., Lin, S., Bingham, S. E., and Webber, A. N.: Bidirectional electron transfer in photosystem I: accumulation of A0- in A-side or B-side mutants of the axial ligand to chlorophyll A0, Biochemistry, 43, 1369–1375, https://doi.org/10.1021/bi0354177, 2004.
Redding, K. E., Luthra, R., Slavov, C., Müller, M., Bullock, B., Holzwarth, A. R., and Rappaport, F.: Directionality of electron transfer within photosystem I, in: 14th International Congress of Photosynthesis, 22–27 July 2007, Glasgow, UK, Photosynth. Res., 91, p. 139, 2007.
Roy, E., Diller, A., Alia, Gast, P., van Gorkom, H. J., de Groot, Huub J M, Jeschke, G., and Matysik, J.: Magnetic field dependence of 13C photo-CIDNP MAS NMR in plant photosystems I and II, Appl. Magn. Reson., 31, 193–204, 2007.
Roy, E., Alia, A., Gast, P., van Gorkom, H. J., Jeschke, G., and Matysik, J.: 13C photo-CIDNP MAS NMR on the reaction center of the green sulphur bacterium at two different magnetic fields, in: Energy from the sun, edited by: Allen, J., Gantt, E., Golbeck, J., and Osmond, B., Springer, Dordrecht, 173–176, 2008.
Sai Sankar Gupta, K. B., Daviso, E., Jeschke, G., Alia, A., Ernst, M., and Matysik, J.: Spectral editing through laser-flash excitation in two-dimensional photo-CIDNP MAS NMR experiments, J. Magn. Reson., 246, 9–17, https://doi.org/10.1016/j.jmr.2014.06.007, 2014.
Saito, K., Shen, J.-R., Ishida, T., and Ishikita, H.: Short hydrogen bond between redox-active tyrosine Y(Z) and D1-His190 in the photosystem II crystal structure, Biochemistry, 50, 9836–9844, https://doi.org/10.1021/bi201366j, 2011.
Santabarbara, S., Galuppini, L., and Casazza, A. P.: Bidirectional electron transfer in the reaction centre of photosystem I, J. Integr. Plant. Biol., 52, 735–749, https://doi.org/10.1111/j.1744-7909.2010.00977.x, 2010.
Scheller, H. V., Jensen, P. E., Haldrup, A., Lunde, C., and Knoetzel, J.: Role of subunits in eukaryotic Photosystem I, Biochim. Biophys. Ac., 1507, 41–60, https://doi.org/10.1016/s0005-2728(01)00196-7, 2001.
Schmid, V. H., Cammarata, K. V., Bruns, B. U., and Schmidt, G. W.: In vitro reconstitution of the photosystem I light-harvesting complex LHCI-730: heterodimerization is required for antenna pigment organization, P. Natl. Acad. Sci. USA, 94, 7667–7672, https://doi.org/10.1073/pnas.94.14.7667, 1997.
Schulten, E. A. M., Matysik, J., Alia, Kiihne, S., Raap, J., Lugtenburg, J., Gast, P., Hoff, A. J., and de Groot, H. J. M.: 13C MAS NMR and photo-CIDNP reveal a pronounced asymmetry in the electronic ground state of the special pair of Rhodobacter sphaeroides reaction centers, Biochemistry, 41, 8708–8717, https://doi.org/10.1021/bi025608u, 2002.
Sosnovsky, D. V., Jeschke, G., Matysik, J., Vieth, H.-M., and Ivanov, K. L.: Level crossing analysis of chemically induced dynamic nuclear polarization: Towards a common description of liquid-state and solid-state cases, J. Chem. Phys., 144, 144202, https://doi.org/10.1063/1.4945341, 2016.
Sosnovsky, D. V., Lukzen, N. N., Vieth, H.-M., Jeschke, G., Gräsing, D., Bielytskyi, P., Matysik, J., and Ivanov, K. L.: Magnetic field and orientation dependence of solid-state CIDNP, J. Chem. Phys., 150, 94105, https://doi.org/10.1063/1.5077078, 2019.
Srinivasan, N. and Golbeck, J. H.: Protein-cofactor interactions in bioenergetic complexes: the role of the A1A and A1B phylloquinones in Photosystem I, Biochim. Biophys. Ac., 1787, 1057–1088, https://doi.org/10.1016/j.bbabio.2009.04.010, 2009.
Thamarath, S. S., Heberle, J., Hore, P. J., Kottke, T., and Matysik, J.: Solid-state photo-CIDNP effect observed in phototropin LOV1-C57S by 13C magic-angle spinning NMR spectroscopy, J. Am. Chem. Soc., 132, 15542–15543, https://doi.org/10.1021/ja1082969, 2010.
Thamarath, S. S., Bode, B. E., Prakash, S., Sai Sankar Gupta, Karthick Babu, Alia, A., Jeschke, G., and Matysik, J.: Electron spin density distribution in the special pair triplet of Rhodobacter sphaeroides R26 revealed by magnetic field dependence of the solid-state photo-CIDNP effect, J. Am. Chem. Soc., 134, 5921–5930, https://doi.org/10.1021/ja2117377, 2012a.
Thamarath, S. S., Alia, A., Daviso, E., Mance, D., Golbeck, J. H., and Matysik, J.: Whole cell nuclear magnetic resonance characterization of two photochemically active states of the photosynthetic reaction center in heliobacteria, Biochemistry, 51, 5763–5773, https://doi.org/10.1021/bi300468y, 2012b.
van Lenthe, E. and Baerends, E. J.: Optimized Slater-type basis sets for the elements 1-118, J. Comput. Chem., 24, 1142–1156, https://doi.org/10.1002/jcc.10255, 2003.
Velde, G. T., Bickelhaupt, F. M., Baerends, E. J., Fonseca Guerra, C., van Gisbergen, S. J. A., Snijders, J. G., and Ziegler, T.: Chemistry with ADF, J. Comput. Chem., 22, 931–967, 2001.
Watanabe, T., Kobayashi, M., Hongu, A., Nakazato, M., Hiyama, T., and Murata, N.: Evidence that a chlorophyll a' dimer constitutes the photochemical reaction centre 1 (P700) in photosynthetic apparatus, FEBS Lett., 191, 252–256, https://doi.org/10.1016/0014-5793(85)80019-3, 1985.
Wawrzyniak, P. K., Beerepoot, M. T. P., de Groot, H. J. M., and Buda, F.: Acetyl group orientation modulates the electronic ground-state asymmetry of the special pair in purple bacterial reaction centers, Phys. Chem. Chem. Phys., 13, 10270–10279, https://doi.org/10.1039/C1CP20213H, 2011.
Webber, A. N. and Lubitz, W.: P700: The primary electron donor of photosystem I, BBA-Bioenergetics, 1507, 61–79, https://doi.org/10.1016/S0005-2728(01)00198-0, 2001.
Wientjes, E., Oostergetel, G. T., Jansson, S., Boekema, E. J., and Croce, R.: The role of Lhca complexes in the supramolecular organization of higher plant photosystem I, J. Biol. Chem., 284, 7803–7810, https://doi.org/10.1074/jbc.M808395200, 2009.
Zill, J. C., Kansy, M., Goss, R., Köhler, L., Alia, A., Wilhelm, C., and Matysik, J.: Photo-CIDNP in the reaction center of the diatom Cyclotella meneghiniana observed by 13C MAS NMR, Z. Phys. Chem., 231, 13288, https://doi.org/10.1515/zpch-2016-0806, 2017.
Zill, J. C., Kansy, M., Goss, R., Alia, A., Wilhelm, C., and Matysik, J.: 15N photo-CIDNP MAS NMR on both photosystems and magnetic field-dependent 13C photo-CIDNP MAS NMR in photosystem II of the diatom Phaeodactylum tricornutum, Photosynth. Res., 140, 151–171, https://doi.org/10.1007/s11120-018-0578-9, 2019.
Zysmilich, M. G. and McDermott, A.: Photochemically induced dynamic nuclear polarization in the solid-state 15N spectra of reaction centers from photosynthetic bacteria Rhodobacter sphaeroides R-26, J. Am. Chem. Soc., 116, 8362–8363, https://doi.org/10.1021/ja00097a052, 1994.